多巴反應性肌張力障礙
多巴反應性肌張力障礙
多徠巴反應性肌張力障礙是指一種好發於兒童或青少年的以肌張力障礙或步態異常為首發癥狀的少見的遺傳性疾病,又稱 Segawa病。該病半數呈散發性,半數呈常染色體顯性遺傳。目前認為,GTP環化水解酶的同工酶GCHⅠ缺乏導致多巴胺合成減少是本病的主要病因。國外學者發現,60%~70%的患者出現GCHⅠ編碼區的基因突變,導致黑質紋狀體系統多巴能神經元GCHⅠ的缺乏,必然導致酪氨酸羥化酶合成減少,最終致多巴胺水平降低。臨床表現為癥狀的晝夜波動性,肢體肌張力異常、震顫、步態怪異等為首發癥狀。治療以小劑量的多巴胺的治療有效。本病需長期用藥,補充多巴胺的不足,堅持用藥治療,預后較好。
● 神經內科、兒科
● 本病半數呈散發性,半數為常染色體顯性遺傳病。主要病因為GTP環化水解酶的同工酶GCHⅠ缺乏導致多巴胺合成減少。故持續給予外源性多巴製劑,可緩解癥狀。
● 國外學者發現,60%~70%的患者出現GCHⅠ編碼區的基因突變位置在14q32.1。有研究提示該病多巴脫羧酶及多巴胺受體是正常的,故持續給予少量外源性多巴製劑,可彌補多巴胺不足,緩解癥狀。
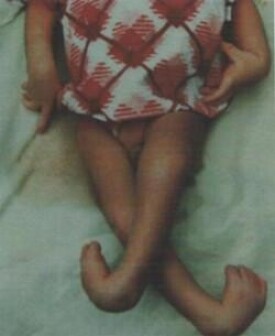
● 多巴反應性肌張力障礙常見於兒童期,女性多於男性。發病年齡一般在4~8歲,但可以早至嬰兒期,晚至成人期。初始癥狀往往是馬蹄內翻足和由於下肢肌張力不全造成的步態異常。以後病情進行性加重,可以出現四肢僵硬,運動徐緩,面無表情。
● 兒童
● ● 發病時首發癥狀常為一側下肢肌張力異常,患兒出現步形怪異、下肢僵硬、步態不穩、馬蹄內翻足等癥狀。有時僅表現為走路較晚,易跌倒。隨病情發展,肌張力異常可影響其他肢體,頭頸部及身體中軸甚至出現痙攣性斜頸、扭轉痙攣。患兒有肢體震顫、肌強直、Babinske征陽性、語言及智能常不受累。
● 成人
● ● 患者發病時以肢體不自主震顫、僵硬等類似帕金森綜合征表現多見。行動遲緩,易疲勞,肢體肌張力高,腱反射亢進,病理呈陽性。大部分患者癥狀有晝夜波動性,晨起或休息后明顯減輕甚而消失,午後或勞累后癥狀可加重。
● 多數患者
● ● 病程呈進行性發展,未經治療者,最終生活不能自理。對小劑量左旋多巴有戲劇性和持久性反應是其顯著的臨床特徵。用藥后所有癥狀包括易疲勞、肌張力障礙、姿勢異常、震顫都會完全消失。
● 確診多巴反應性肌張力障礙需行腦電圖、顱腦影像學檢查等排除其他疾病,必要時進行分子基因檢測。
● 血、尿、便常規,通常正常。
● 腦脊液檢查可正常,腦脊液中高香草酸及生物喋呤含量可降低。
● 肝功能檢查正常,有鑒別診斷意義。
● 腦電圖、誘發電位、顱腦CT、MR及PET檢查,均正常。
● 針對三種基因(GTPCH、SR、TH)的多基因面板測試。
● 根據兒童或成人起病時,以原因不明的肢體肌張力異常、震顫、步態怪異等為首發癥狀,晨輕暮重為主要臨床特點,尤其有家族遺傳史者,且對小劑量多巴製劑有療效,應高度懷疑本病。
● 一些其他疾病也可出現運動遲緩、震顫、肌張力增高等癥狀,須注意與多巴反應性肌張力障礙相鑒別。這些疾病包括腦性癱瘓、少年型帕金森病、肝豆狀核變性、痙攣性截癱等。
● 一旦出現上述癥狀,需要及時去醫院就診,進行詳細檢查,請醫生進行診斷和治療。
● 小劑量多巴製劑對本病療效。半數患者用藥當天見效,起效時間一般不超過7日。一旦懷疑本病,立即用藥,可為診斷性治療。
● 左旋多巴/苄絲肼(美多巴)可長期持續用藥,副作用少;若停葯,癥狀即再出現。
● 多巴反應性肌張力障礙如不治療,可引起患兒走路不穩,易摔倒,導致骨折。還可出現痙攣性斜頸、扭轉痙攣等癥狀。
● 多巴反應性肌張力障礙的病因為GCHI基因突變致多巴胺合成減少,需長期補充,不需增加劑量,預后較好。
● 近親避免結婚、遺傳諮詢、攜帶者基因檢測、產前診斷和選擇性人工流產等,防止患兒出生。
● 早期診斷,早期治療,加強臨床護理,對改善患者的生活質量有重要意義。
基本信息
- 中文名
- 多巴反應性肌張力障礙
- 外文名
- dopa-reactivedystonia
- 別名
- 肌張力障礙
- 傳播途徑
- 遺傳因素
- 癥狀表現
- 肢體肌張力異常、震顫、步態怪異等為首發癥狀,晨輕暮重為主要特點
- 就診科室
- 精神病科
- 原因
- GTP環化水解酶的同工酶GCHⅠ缺乏導致多巴胺合成減少。
- 多發人群
- 兒童或青少年