戈謝病
戈謝病
戈謝病是一種家族性糖脂代謝病。為染色體隱性遺傳,是溶酶體沉積病中最常見的一種。本病罕見,我國在1948年首次報道以來,各地均有報告,尤其河北、山東、河南及遼寧病例報告較多。本病的發生與基因變異有關。相關基因變異,會導致β-葡糖腦苷脂酶缺乏,造成葡萄糖腦苷脂在肝、脾、肺、腎、骨和中樞神經系統的細胞的中蓄積,從而導致這些器官發生病變。根據神經系統是否受累,將本病分為3個亞型。Ⅰ型(非神經病變型):常表現為面色蒼白、全身無力、皮膚及牙齦出血、月經量增多等。Ⅱ型(暴發性神經病變型):面色蒼白、全身無力、癲癇發作、認知障礙等。Ⅲ型(慢性或亞急性神經病變型):早期與Ⅰ型相似,逐漸出現神經系統受累表現,如走路不穩、動作不協調、精神錯亂等。治療方法包括:對症治療、脾切除、酶替代治療、基因治療、其他治療。部分患者可能會出現脾梗死、脾破裂、嚴重的骨病、出血併發症、感染、肝衰竭或嚴重肺疾病等併發症。這些併發症可導致患者死亡。戈謝病Ⅰ型進展緩慢,脾切除后可長期存活,智力正常,對β-葡糖腦苷脂酶製劑替代治療反應顯著,預后最好。戈謝病Ⅱ型多於發病後1年內死於繼發性感染,少數患者可存活2年以上。戈謝病Ⅲ型多由於神經系統癥狀較重,死於併發症。
● 血液科或內科
● 戈謝病是一種罕見的常染色體隱性遺傳病,其發生與基因變異有關。
● 根據神經系統是否受累,將本病分為3個亞型。
● 較多見,起病慢,約2/3患者在兒童期發病,發病越早,癥狀越重。
● 本型會引起血小板減少和貧血,常表現為面色蒼白、全身無力、皮膚及牙齦出血、月經量增多等。
● 多數患者還會出現骨骼受累,表現為骨痛。
● 部分患者肺部受累,可出現氣促、咳嗽、呼吸困難等。
● 罕見,常在新生兒期及嬰兒期發病,病情進展快。
● 血小板減少、貧血等表現與Ⅰ型相似。
● 本型會快速出現神經系統癥狀,如延髓麻痹(聲音嘶啞、失聲、進食困難等)、癲癇發作、角弓反張(身體後仰,像弓箭一樣),以及認知障礙等。
● 重度患者還可能會出現關節畸形、關節活動障礙。
● 常發病於兒童期,進展緩慢。
● 早期表現與Ⅰ型相似,逐漸出現神經系統受累表現,如走路不穩、動作不協調、精神錯亂等。
● 血常規、β-葡糖腦苷脂酶活性測定、殼三糖酶活性檢測、基因檢測、骨髓細胞學檢查、影像學檢查等檢查,可以幫助醫生診斷疾病。
● 本病常導致患者血小板減少和貧血,通過血常規檢查可以幫助醫生判斷病情的嚴重程度。
● β-葡糖腦苷脂酶活性測定是診斷戈謝病的金標準。
● 當其外周血白細胞或皮膚成纖維細胞中葡糖腦苷脂酶活性降低至正常值的30%以下時,即可確診。
● 但少數患者酶活性減低不到此標準,則需檢測血漿殼三糖酶活性,並進一步做基因突變檢測確診。
● 戈謝病患者血漿殼三糖酶活性,與正常人相比,顯著升高。通過對其進行檢測,有利於確診疾病。
● 通過基因突變型的分析可助診斷。
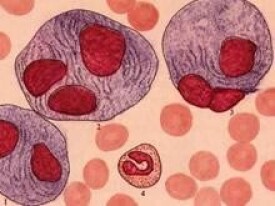
● 如果在骨髓中發現戈謝細胞時,醫生會高度懷疑戈謝病,但並不能確診本病,還需要排除其他疾病,同時還要進行β-葡糖腦苷脂酶活性測定。
● B 超、CT等影像學檢查,可以發現腫大的肝臟、脾臟,有助於醫生判斷病情嚴重程度。
● 醫生根據臨床表現、β-葡糖腦苷脂酶活性測定、骨髓細胞學檢查、影像學檢查等可以進行診斷。
● 出現面色蒼白、全身無力、皮膚及牙齦出血;骨痛;走路不穩、動作不協調、精神錯亂等表現。
● β-葡糖腦苷脂酶活性測定,發現外周血白細胞或皮膚成纖維細胞中葡萄糖腦苷脂酶活性降低。
● 骨髓塗片檢查見到戈謝細胞。
● 影像學檢查發現肝、脾腫大。
● 戈謝病,需要與慢性粒細胞白血病、多發性骨髓瘤、骨髓增生異常綜合征、地中海貧血、肺結核等疾病相鑒別。
● 如果出現面色蒼白、全身無力、皮膚及牙齦出血,或者骨痛、動作不協調時,應及時到醫院掛號就診。
● 醫生通過β-葡糖腦苷脂酶活性測定、骨髓細胞學檢查、影像學檢查等可以進行診斷和鑒別診斷。
● 治療方法包括:對症治療、脾切除、酶替代治療、基因治療、其他治療。
● 貧血者平時可以補充維生素及鐵劑,必要時可進行成分輸血,以糾正貧血或血小板減少。
● 發生骨骼病變者,可通過止痛、理療、人工關節置換等方式治療,還可使用鈣劑和雙磷酸鹽治療骨質疏鬆。
● 適用於巨脾,伴脾功能亢進者,年齡在4~5歲或5歲以上,可以改善癥狀。
● 對於Ⅰ型和Ⅲ型部分患者建議脾切除術。
● 注射用伊米苷酶替代治療,對於延長患者壽命、提高生存質量有顯著效果。絕大多數患者的癥狀改善,臟器不再繼續受累。
● 主要用於戈謝病Ⅰ型治療。
● 其他治療,如底物減少療法、分子伴侶療法、基因治療等方法均在探索中。
● 戈謝病可造成骨骼、消化、呼吸、中樞中心等系統的損害,嚴重者可危及生命。
● 部分患者可能會出現脾梗死、脾破裂、嚴重的骨病、出血併發症、感染、肝衰竭或嚴重肺疾病等併發症。這些併發症可導致患者死亡。
● 戈謝病Ⅰ型進展緩慢,脾切除后可長期存活,智力正常,但生長發育落後。對葡糖腦苷脂酶製劑替代治療反應顯著,預后最好。
● 戈謝病Ⅱ型多於發病後1年內死於繼發性感染,少數患者可存活2年以上。
● 戈謝病Ⅲ型多由於神經系統癥狀較重,死於併發症。
● 產前診斷是預防本病發生的有效措施。進行產前診斷,有目的地檢查胎兒是否存在某種酶或某種基因缺陷,可以在產前明確胎兒是否患病,以採取措施保證優生。
基本信息
- 中文名
- 戈謝病
- 外文名
- Gauchers disease
- 別名
- 家族性脾性貧血、腦甙病、腦苷脂網狀內皮細胞病
- 癥狀表現
- 出血傾向 抽搐 吞咽困難
- 就診科室
- 內科
- 原因
- 常染色體隱性遺傳
- 併發症
- 脾破裂 骨折
- 是否傳染病
- 否
- 季節分佈
- 四季