Silver-Russell綜合征
Silver-Russell綜合征
早在1953及1954年,由Silver醫師及Russell醫師分別報告了一群在子宮內生長遲滯( IUGR,intrauterine growth retardation )的兒童,同時具有身材矮小、臉小呈三角形、低位耳、第五手指彎曲等特徵的疾病,因此命名為Russell-Silver syndrome。Russell-Silve綜合征(簡稱RSS)又稱為Silver-Russells綜合征或Silver綜合征。RSS是一種罕見的先天疾病,主要特徵為身材矮小,且通常在身體二側呈現左右不對稱。 2018年5月11日,該疾病被列入國家衛生健康委員會等5部門聯合制定的《第一批罕見病目錄》。
致病原因並不單純,病因學並不明了,基本上認為與遺傳有關,但是致病基因並不清楚。在病因學上RSS有多種遺傳模式,通常沒有家族史,普遍上均是家族中的單一案例。估計有7-10%的RSS患者是因為第7對染色體母源單親二體症(maternal uniparentaldisomy),另有少於1%的RSS患者在染色體17q25處有重組現象。RSS的分子遺傳機制仍未明朗,幾個零星個案顯示可能和位於第七對染色體上7p11.2– p12區域間一個GRB10 (human growth factorreceptor-bound protein 10)的基因有關。主要有下列三種遺傳模式:
(1) 兩個第七對染色體均來自於母親,即所謂的母源單親二體症(maternal uniparental disomy,簡稱UPD),大約10%的案例屬於這種遺傳模式。但絕大多數均為偶發性,幾乎所有案例均是家族中唯一的個案。
若屬於此種模式,則家族成員發生之機率如下:
(a) 個案之父母:父母雙方均未患病。
(b)個案之兄弟姊妹:兄弟姊妹發病之機率與一般人無異。
(c) 個案之子代:子代再發之機率不高。
(d) 其他之家族成員:發病之機率與一般人無異。
(2)體染色體顯性遺傳,但非常罕見。這種模式系成對染色體中二個基因任一個突變即會致病,若父母親任何一人患病,則子代有二分之一的機會也會患病,二分之一的機會則可以生出正常的子代。
(3) 體染色體隱性遺傳,也是非常罕見。這種模式系成對染色體中二個基因均突變才會致病,若父母親任何一人帶有此致病基因,則子代有二分之一的機會也會帶有此突變基因,但不會患病,四分之一可以生出正常的小孩,四分之一的機會則會生出患病的子代。
除此之外的個案則是多重原因所造成。在詢問家族史時必須包括父母及兄弟姊妹出生時的身高體重,若家族中均無其他人患病,則均屬偶發性。
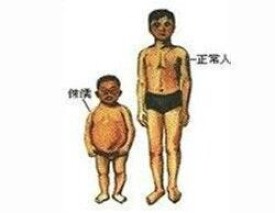
此病有三個臨床特徵: .子宮內出現身材矮小、兩側骨骼不對稱以及小指短且內彎。
生長遲緩源自出生前的子宮內生長遲滯( intrauterine growth retardation, IUGR ),出生后也是生長不良( failure to thrive )。典型RSS患者會身材矮小,出生時的體重會比平均值低2個標準偏差,出生后的體重及身高也會平均值低2個標準偏差。
因骨骼發育不對稱,患者可能有頭部、軀幹與四肢骨骼的左右不對稱。其中以四肢最明顯。
患者有異常的臉部特徵,臉小呈三角形使前額顯得突出,或因小臉顯得上半部的頭比較大,下巴偏小。但頭圍都在正常範圍內。
其他可能發現的狀況:
在嬰兒及幼兒時骨骼發展不成熟,前囪門較晚閉合、藍鞏膜、易出汗,尤其頭與上半身;胃食道逆流。肌肉張力較低。男嬰可能有隱睾。第二和第三足並指,脊椎異常,薦骨尾骨缺乏。皮膚會有咖啡牛奶斑。禁食過久易低血糖(尤其10個月大至兩三歲間更明顯)青春期會提早,患童可能有發展遲緩(包括運動及認知)及學習障礙,但智能正常。其他: 如先天性心臟病、泌尿系統異常(腎臟結構異常、后尿道瓣、尿道下裂。腫瘤(顱咽管瘤、精原細胞瘤、肝癌和Wilms瘤)。
所以診斷原則上是基於臨床特徵為主:出生時體重比平均值低2個標準偏差;出生後生長的體重及身高比平均值低2個標準偏差;成比例的身材矮小,骨齡發展遲緩;頭圍正常,常有pseudohydrocephalus現象;典型的臉部特徵如寬顯的前額、小且三角形臉、下巴小窄;四肢長度不平衡;其他有助於診斷的特徵,如第五手指彎斜、並指、咖啡牛奶斑、二手臂長度較身高短。
並無特別的實驗室檢測方法可供診斷RSS,尤其是因為此疾病之病因學異質性高,可以進行下列之檢查:細胞遺傳學檢查-有些RSS患者在染色體17q25處有重組的現象。放射線骨骼檢查-要排除骨骼的發育不良而造成類似RSS現象,可由X光檢查骨齡發展是否遲緩。分子遺傳學檢查-RSS的基因或相關基因至目前尚未知,單一親源二體症的研究有助於建立第7對染色體母源單親二體症的診斷,而大部份RSS患者對於他們的診斷並無可供檢定的原因。
對於此病並無特殊治療法,只能給予癥狀治療。治療主要系著重於身高及生長,仔細地觀察及注意生長速率是很必需的。對於身高方面,可考慮給予生長激素治療,但無法改善肢體長短不均的現象。兒童期給與生長激素仍不易達到正常身高,成人後看起來幾近正常。
對於腳長不一可以用架治療以預防問題發生。下肢長短腳情形若超過75px以上,則必須進行治療,否則會導致代償性的(即非原發性的)脊柱側彎。初始的治療可穿加高跟的鞋子,年紀稍長時,下肢長度不一致的情形可藉由骨延長術(distraction osteogenesis)或是生長板融合術(epiphysiodesis)來矯正。
在六個月至三歲間易發生空腹低血糖,故這期間若有生病時,應多吃與攝取足夠葡萄糖。低血糖現象是在治療上一個重要的課題,RSS的病童皮下脂肪較少,通常都胃口不佳而且比較瘦,所以長時間的治療例如動手術等會有比較高的危險性產生急性低血糖。
如合併有顱顏異常,可在孩童時期經由小兒牙醫來治療,或是於成人時期施行牙齒矯正。對於某些較嚴重的唇顎裂或下顎過小的病人,則建議應由一個全方位的顱顏手術專家小組來進行治療。
胃腸部分的異常則需加以積極治療,需以鋇劑攝影、內視鏡及酸鹼度(pH)測定等方式來評估食道炎之情形。建議以防止食道胃酸逆流(gastroesophageal reflux)的餵食姿勢或喂以較濃稠的食物。避免高油脂食物如巧克力、咖啡、可樂等。若傳統保守療法不見成效時則需施以制酸劑(通常會使用氫離子泵浦抑製劑,例如omeprazole或patoprazole),目的在減少胃酸分泌。比較嚴重的個案則必須施行手術例如胃底折迭術(fundoplication)來加以改善。
嬰孩時期若出低張力現象則會有發育遲緩的可能性,此時早療系統及物理治療的介入是必要的。同時必須小心地觀察是否有語言上說跟聽的問題,若有任何遲緩現象,也要快速地求助於聽語治療。
因為身體上任何的異常或較為矮小也都會造成孩童對自我身體形象的敏感,這些因素在其自信心、同儕關係及社交技巧上扮演很重要的角色。因此適當的心理咨商將有助於孩童的成長。
兒童期雖常纖瘦,體重不足,肌肉無力,發展遲緩,學習障礙,但青春期后漸改善,還是可以成為一個健康的人,但成人還是比一般人稍矮;而此病並不會造成智障,仍有正常的智力。
1.RSS有多重病因學,大部份家族只有出現一個患者,已知的病因包括第7對染色體源自母親二體症(maternaluniparental disomy)及體染色體顯性遺傳或體染色體隱性遺傳。
2.當一個RSS患者是因第7對染色體源自母親二體症時,可預知父母親一定沒有患此病,而患者手足患病之風險並不會增加,患者之下一代的風險可能很低。
3.產前診斷對於RSS通常不可行。但若是屬於第七對染色體的母源單親二體症(maternal UPD7)之個案,經由羊膜穿刺進行小孩及父母之。產前分子檢測是可行的。而若要在子宮內觀察是否有生長遲緩現象需使用超音波來檢查。一般而言,要能清楚仔細地看到生長發育情形,通常得等到懷孕後期(約28周時),因此也造成產前診斷的限制性。
4.對於家族中只有單一患者的情況,大部份的懷孕並不會增加RSS的風險。
基本信息
- 中文名
- Silver-Russell綜合征
- 別名
- Silver-Russell矮小症