腎上腺腦白質營養不良
腎上腺腦白質營養不良
腎上腺腦白質營養不良是以X性連鎖隱性遺傳為主要遺傳方式的脂質代謝異常性疾病。半數以上患者於兒童或青少年期起病,發病率為(0.5~1)/10萬,95%為男性。無種族和地域特異性。此病多屬於X性連鎖隱性遺傳。由於溶酶體過氧化物酶的缺乏,導致極長鏈脂肪酸在細胞內異常沉積,特別是在腎上腺皮質和腦白質內沉積,導致腎上腺和腦白質破壞。主要表現為進行性精神運動障礙,如出現多動症、注意力下降、攻擊行為、步態不穩、癱瘓,以及視力、聽力下降和(或)腎上腺皮質功能減退等,如皮膚色素沉著、無力等。本病無特效治療,以對症治療為主,可以延緩病情進展。本病預后差,在發病後2~4年內病情呈進行性惡化直至死亡,一般不超過9年。
● 神經內科、內分泌科或內科
● 腎上腺腦白質營養不良為基因遺傳性疾病。
● 有疾病家族史,兒童或青少年期發病,常為X性連鎖隱性遺傳。
● 新生兒型為常染色體隱性遺傳。
● 本病主要表現為進行性精神運動障礙,視力及聽力下降和(或)腎上腺皮質功能減退等。分為兒童腦型、青少年腦型、成人腦型、腎上腺脊髓神經病型、Addison型、無癥狀型、雜合子型。
● 兒童腦型
● ● 初期表現為注意力不集中、記憶力減退、學習困難、走路不穩、行為異常等。
● ● 逐漸出現視力和(或)聽力下降、發音障礙、癱瘓、癲癇發作、痴獃等癥狀。
● ● 病情逐步進展,最終完全癱瘓,失明或耳聾,可有驚厥,甚至出現驚厥持續狀態。
● 青少年腦型
● ● 臨床表現類似於兒童腦型,但進展緩慢。
● 成人腦型
● ● 多於21歲以後起病,腦內迅速進展,臨床表現類似於兒童腦型。
● 腎上腺脊髓神經病型
● ● 表現為進行性下肢痙攣癱瘓,大、小便功能障礙和性功能障礙等,癱瘓進展緩慢,可伴周圍神經損害,出現步態不穩、精神行為異常、智力減退等,有腎上腺皮質功能減退表現,如皮膚色素沉著、無力等。
● Addison型
● ● 表現為原發腎上腺皮質功能減退,臨床可見皮膚髮黑、嗜鹽、多汗、疲乏無力、經常嘔吐、腹瀉、暈厥等。
● 無癥狀型
● ● 患者一般無明顯癥狀。
● 雜合子型
● ● 女性雜合子中20%~30%可有輕微的神經系統癥狀,多表現為類似腎上腺脊髓神經病的痙攣性截癱,但癥狀較輕,很少出現腦部癥狀、周圍神經病及腎上腺功能減退的癥狀。
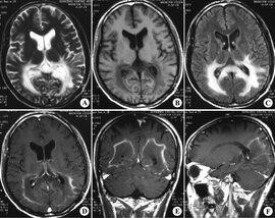
● 確診腎上腺腦白質營養不良需要做血漿極長鏈脂肪酸測定、磁共振成像、CT及基因監測等。
● 血漿極長鏈脂肪酸測定
● ● 血漿極長鏈脂肪酸異常升高對診斷腎上腺腦白質營養不良具有重要價值。
● CT和磁共振成像
● ● 可發現本病所特有的腦部表現,為疾病診斷提供重要線索。
● 基因檢測
● ● 可發現基因突變,明確診斷。
● 醫生根據病史、臨床表現、輔助檢查診斷腎上腺腦白質營養不良。
● 病史
● ● 患者多為男性,兒童或青少年起病。
● 臨床表現
● ● 步態不穩、行為異常、偏癱、皮質盲、耳聾等,且緩慢進行性加重。
● 輔助檢查
● ● CT或MRI表現為兩側腦室三角區周圍白質內和枕葉大片對稱性低密度區或信號異常區,病灶可向兩額部伸展,可有病灶邊緣的強化。
● ● 血漿極長鏈脂肪酸濃度增高具有診斷價值,ABCD1基因異常可以確診。
● 腎上腺腦白質營養不良需要與其他類型腦白質營養不良、Schilder病等相鑒別。
● 如果出現上述類似的癥狀,需要及時去醫院就診。
● 醫生通過家族史、臨床表現及輔助檢查進行診斷及鑒別診斷。
● 本病尚無特效治療,多種方法正在探索之中,如飲食治療、激素替代治療、基因治療等。
● 藥物治療
● ● 臨床上出現腎上腺功能減退者,可使用糖皮質激素替代治療。
● ● 洛伐他汀可降低患者皮膚或成纖維細胞中極長鏈脂肪酸的水平。
● 手術治療
● ● 骨髓移植。少數病例經過此項治療,可以穩定臨床癥狀。
● 其他治療
● ● 飲食治療、康復訓練、基因治療、造血幹細胞移植等。
● 本病可嚴重影響患者的日常生活及工作,患者病情日漸加重,給家庭、社會帶來較大的負擔。且病情進展快,預后不良。
● 本病治療方法以對症治療為主,糖皮質激素可延長生命,部分緩解神經系統癥狀,但不能阻止髓鞘破壞和改變病程進展。預后較差,通常在9年內進展至死亡。
● 限定飲食中極長鏈脂肪酸的攝入,進食Lorenzo油可減少極長鏈脂肪酸的合成。
● 增加睡眠與休息,增強抵抗力,避免感染。
● 做好孕前檢查與遺傳諮詢工作,有利於優生優育。
基本信息
- 中文名
- 腎上腺腦白質營養不良
- 外文名
- adrenoleukodystrophy,ALD
- 原因
- 遺傳
- 所屬科室
- 內科 - 神經內科