假肥大型肌營養不良
假肥大型肌營養不良
假肥大型肌營養不良是以進行性骨骼肌無力為特徵的一組原發性骨骼肌壞死性疾病。是原發於肌肉組織的X連鎖隱性遺傳的肌病。發病機制不明確。主要表現為兒童期發病,走路易跌,奔跑困難、急性胃擴張、智能遲緩、認知功能下降、學習困難、癲癇,還會出現情感、行為問題,晚期、反覆感染可出現心力衰竭、心律失常、急性胃擴張、癲癇等。目前尚無特效治療,主要是對症支持治療。可出現心力衰竭、心律失常、急性胃擴張、癲癇等。本病不能根治,經治療可延緩病情的發展,減輕癥狀,延長生命。
● 神經內科或內科
● 為原發於肌肉組織的X連鎖隱性遺傳的肌病,發病機制不明。
● 本型又可分為兩種類型:Duchenne型肌營養不良(DMD)、Becker型肌營養不良(BMD),具體表現如下:
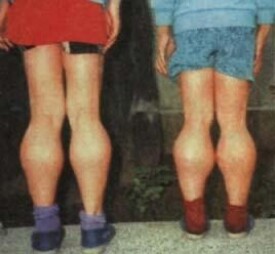
● DMD患者兒童期發病,一般在4~6歲時走路易跌,奔跑困難,逐漸出現走路和上樓困難,下蹲站起困難。神經系統檢查可見四肢肌力低下,肌肉萎縮,腱反射減弱。由於骨盆帶肌肉無力而呈典型的鴨步,肩帶肌肉萎縮無力形成翼狀肩或遊離肩,腹肌和髂腰肌的萎縮無力形成特徵性的Gowers征。絕大多數患兒有腓腸肌假性肥大,少部分可見舌肌或三角肌假性肥大。BMD患者多在5~15歲起病,臨床表現與DMD患者相似。
● 大多數DMD、BMD患者無心血管癥狀,只有在疾病晚期和反覆感染的應激情況下,才出現心力衰竭和心律失常。
● 胃腸道的平滑肌也可受累。急性胃擴張可導致死亡,部分患者可有嚴重便秘。
● DMD和BMD患者,可有中樞神經系統功能障礙尤其是智能遲緩,患者癲癇的發病率增高,尤其是BMD型。DMD患者易出現情感、行為問題,認知功能下降及學習困難。
● 能行走的DMD患者,腰椎骨密度輕度降低;而不能行走者則明顯降低。
● 確診假肥大型肌營養不良還需病理組織學檢查、基因診斷等。
● 肌酸磷酸激酶(CK)明顯升高。血清CK升高可出現於出生時,疾病後期略有降低。
● 特徵性的病理改變有散在的退行性變和壞死肌纖維。隨著時間的延長,出現肌內膜結締組織增加以及肌纖維的喪失,脂肪組織的替代。
● 應用定量PCR測定、短串聯重複序列連鎖分析檢出DMD基因攜帶者。
● 為肌源性改變,病變肌肉呈低電位,波形持續時間縮短,而多相波增高。
● 醫生根據臨床表現和實驗室檢查、基因檢查可以作出診斷。
● 主要表現為兒童期發病,走路易跌,奔跑困難、急性胃擴張、智能遲緩、認知功能下降、學習困難、癲癇等。
● 血清生化檢查示肌酸磷酸激酶(CK)明顯升高。
● 肌肉活組織檢查示有散在的退行性變和壞死肌纖維。
● 肌電圖示肌源性改變。
● 基因檢查可確診。
● 一些疾病也可能出現走路易跌,奔跑困難、智能遲緩、認知功能下降、學習困難、癲癇等。容易與假肥大型肌營養不良相混淆。這些疾病包括強直性肌營養不良、神經性肌強直、僵人綜合征等相鑒別。
● 如果出現上述類似的癥狀,需要及時去醫院就診,進行詳細檢查,請醫生進行診斷和治療。
● 假肥大型肌營養不良主要採取對症支持治療等。
● 常用的藥物有:維生素E、肌苷、三磷腺苷以及中藥等。但上述治療只能延緩病情的發展,並不能根本治癒疾病。
● 為保持肌肉功能及預防攣縮,進行適度運動甚為重要,不宜久卧床上。對症治療包括肌肉、關節被動運動和按摩,注意並預防併發症。
● DMD患者常發展為進行性脊柱側彎,常需行脊柱后融合術。
● 可出現心力衰竭、心律失常、急性胃擴張、癲癇等。
● 急性胃擴張可導致死亡。
● 本病不能根治,經治療可延緩病情的發展,減輕癥狀,延長生命。
● 做好遺傳諮詢,通過家系調查、CK測定,DNA分析,以及對已懷孕的基因攜帶者進行胎兒產前診斷,防止患兒出生。
基本信息
- 中文名
- 假肥大型肌營養不良
- 外文名
- Duchenne Muscular Dystrophy, DMD
- 是否遺傳
- 是
- 多發人群
- 兒童
- 就診科室
- 神經內科
- 癥狀表現
- 走路易跌,奔跑困難,等
- 常見發病
- 腓腸肌