過渡態理論
A.G.埃文斯等提出的理論
渡態理論即活化絡合物理論,(transition-state theory)。過渡態:以量子力學對反應過程中的能量變化的研究為依據,認為從反應物到生成物之間形成了勢能較高的活化絡合物,活化絡合物所處的狀態叫過渡態。過渡態理論是1935年由A.G.埃文斯和M.波拉尼提出的,研究有機反應中由反應物到產物的過程中過渡態的理論。
目錄
研究有機反應中由反應物到產物的過程中過渡態的理論。過渡態理論是由A.G.埃文斯和M.波拉尼於1935年提出的。
過渡態和活性中間體 有機反應可分為一步反應(協同反應)和分步反應兩大類。其中,一步反應只有過渡態,沒有活性中間體;而分步反應既有過渡態,又有活性中間體。活性中間體並非就是過渡態,兩者不可混淆。例如氯代叔丁烷的水解反應分二步進行:
其間出現兩個過渡態、一個中間體──正碳離子。這兩步反應分別經過兩個勢壘和,過渡態1和過渡態2和分別出現於每步的勢能頂峰處,而活性中間體處於兩峰之間的凹谷處(圖1)。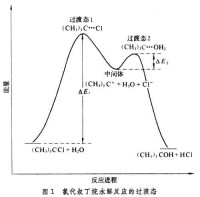
過渡態理論
活性中間體與兩個過渡態的結構和性質相近,但不相同。一般來說,中間體很活潑,壽命很短,但比過渡態要穩定,故可用各種現代物理化學方法測定其結構。兩個過渡態之間的中間體的勢能越低,則中間體越穩定。利用中間體結構和性能的知識,可大致推斷出過渡態的結構、性能,以闡明反應機理。
反應速率決定步聚和產物決定步聚 由圖1還可以看出,第一步反應的活化能比第二步反應大很多(),因此第一步反應比第二步反應慢得多。第一步反應是總反應速率的關鍵,在化學動力學上稱為反應速率決定步聚,第一步的過渡狀態稱為速率決定過渡態。
如果在上述的氯代叔丁烷水溶液中加入第二種比水的親核性更強的試劑,例如疊氮負離子,則反應的主要產物為叔丁基疊氮化物,而原有產物叔丁醇很少。這是由於水和分別與叔丁基正碳離子反應並相互競爭。對於兩者來說,第一步均形成叔丁基正碳離子,是速率決定步聚,第二步反應是競爭反應。
從第二步反應的過渡態能量圖(圖2)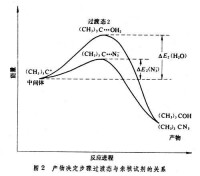 來看,由於的親核性比水強,其第二步反應活化能更小,即,由於與反應要比水快,在第二步反應的競爭中佔了優勢,故主要產物為。結合圖1來看,第二步反應的勢壘)比第一步()小得多,反應速率也高得多,是決定所得產物的步聚,因此稱第二步反應為產物決定步聚。由此可見,如果知道一個有機反應中的中間體和過渡態的類型,並通過動力學方法確定其中的速率決定步驟和產物決定步驟,就能推知整個反應機理。
來看,由於的親核性比水強,其第二步反應活化能更小,即,由於與反應要比水快,在第二步反應的競爭中佔了優勢,故主要產物為。結合圖1來看,第二步反應的勢壘)比第一步()小得多,反應速率也高得多,是決定所得產物的步聚,因此稱第二步反應為產物決定步聚。由此可見,如果知道一個有機反應中的中間體和過渡態的類型,並通過動力學方法確定其中的速率決定步驟和產物決定步驟,就能推知整個反應機理。
過渡態理論
活化熵的意義 由過渡態理論的基本公式
可以知道, 反應活化熵 對反應速率有一定的影響,通常其作用比活化焓 要小,然而從反應活化熵的變化可以獲得有關過渡態立體化學特徵的知識。通常,若 是負值,即熵值減少,則過渡態結構的有序性有所增加;反之,若是正值,則其有序性減少, 負值過大不利於反應。此外, 還必須綜合比較和 變化的相對大小對於活化自由能 的影響。例如,兩端為和基團的10個碳原子的直鏈化合物在關環成內酯時,其過渡態結構的有序性大為增加,熵值減少很多,即上述公式的第二項向正值方向變化,使 向正值方向變化也大,不利於反應;而含3、4個碳原子的化合物在進行相應的關環反應時,雖然熵值負值很小,但正值很大,的變化難以抵消 的不利作用,結果活化能很高,反應也難以發生。五、六元環的和綜合作用最為有利,所以容易成環。
參考書目
J.馬奇著,陶慎熹、趙景旻譯:《高等有機化學》,人民教育出版社,北京,1981。(J.March,Advanced organic Chemistry,2nd ed., McGraw-Hill, New York, 1977.)
D. S. Kemp and F. Vellaccio,Organic Chemistry,Worth Pub., New York, 1980.