活化能
活化能
活化能是指分子從常態轉變為容易發生化學反應的活躍狀態所需要的能量。 (阿倫尼烏斯公式中的活化能區別於由動力學推導出來的活化能,又稱阿倫尼烏斯活化能或經驗活化能)活化分子的平均能量與反應物分子平均能量的差值即為活化能。
活化能是一個化學名詞,又被稱為閾能。這一名詞是由阿倫尼烏斯(Arrhenius)在1889年引入,用來定義一個化學反應的發生所需要克服的能量障礙。活化能可以用於表示一個化學反應發生所需要的最小能量。反應的活化能通常表示為E,單位是千焦耳每摩爾(kJ/mol)。
對一級反應來說,活化能表示勢壘(有時稱為能壘)的高度。活化能的大小可以反映化學反應發生的難易程度。
在Arrhenius提出活化能概念之前,人們對溶液反應曾總結出這樣一個規則:溶液溫度每升高10℃,反應速率將成倍增加。並且,在1878年,由英國科學家Hood最早通過實驗歸納出一經驗關係式:
lgk=B-C/T
式中B、C是經驗常數。
dlnKc/dT= ⊿U/RT^2
並導出了溫度與反應速率常數之間的關係式:
dlnk/dT=(A/RT^2)+I
不過他沒有給出A的物理意義以及確定的 I方法,因此當時沒能引起人們的重視
1889年,Arrhenius 通過大量實驗與理論的論證,揭示了反應速率與溫度的關係Arrhenius經驗公式,其形式如下:
指數式 k=Ae^-Ea/RT
對數式 lnk=lnA-Ea/RT
微分式 dlnKc/dT= ⊿U/RT^2
阿倫尼烏斯提出了活化能的概念,但對活化能的解釋不夠明確,特別是把活化能看作是與溫度無關的常數,這與許多實驗事實不符。 20世紀20年代,科學家托爾曼運用統計熱力學來討論化學反應速率與溫度的關係,並於1925年推導出下面的反應式:
Ea=-
式中:為活化分子的平均摩爾能量,未反應物分子的平均摩爾能量,活化能 是活化分子的平均能量與反應物分子的平均能量之差。
我們可以看出,在上式中,和都與溫度有關,因此Ea也應是溫度的函數,但在有些情況下二者的溫度效應可能彼此抵消,此時活化能則與溫度無關。托爾曼所推導出的公式較好地彌補了阿倫尼烏斯理論的一些不足與缺陷,不再將活化能與溫度相互隔離開來,而是提出了一個更為普遍與更具說服力的一種解釋。
活化能是指化學反應中,由反應物分子到達活化分子所需的最小能量。以酶和底物為例,二者自由狀態下的勢能與二者相結合形成的活化分子的勢能之差就是反應所需的活化能,因此不是說活化能存在於細胞中,而是細胞中的某些能量為反應提供了所需的活化能。
化學反應速率與其活化能的大小密切相關,活化能越低,反應速率越快,因此降低活化能會有效地促進反應的進行。酶通過降低活化能(實際上是通過改變反應途徑的方式降低活化能)來促進一些原本很慢的生化反應得以快速進行(或使一些原本很快的生化反應較慢進行)。影響反應速率的因素分外因與內因:內因主要是參加反應物質的性質;在同一反應中,影響因素是外因,即外界條件,主要有濃度、壓強、溫度、催化劑等。
化學反應的活化能
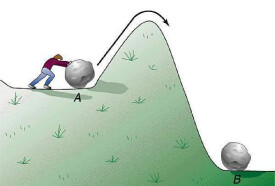
實驗證明,只有發生碰撞的分子的能量等於或超過某一定的能量Ec(可稱為臨界能)時,才可能發生有效碰撞。具有能量大於或等於Ec的分子稱為活化分子。
活化能原理
圖1中,Ea表示分子的平均能量,Ec是活化分子具有的最低能量,能量等於或高於Ec的分子可能產生有效碰撞。活化分子具有的最低能量Ec與分子的平均能量Ea之差叫活化能。
不同的反應具有不同的活化能。反應的活化能越低,則在指定溫度下活化分子數越多,反應就越快。
不同溫度下分子能量分佈是不同的。圖2是不同溫度下分子的能量分佈示意圖。當溫度升高時,氣體分子的運動速率增大,不僅使氣體分子在單位時間內碰撞的次數增加,更重要的是由於氣體分子能量增加,使活化分子百分數增大。圖2中曲線t1表示在t1溫度下的分子能量分佈,曲線t2表示在t2溫度下的分子能量分佈(t2>t1)。溫度為t1時活化分子的多少可由面積A1反映出來;溫度為t2時,活化分子的多少可由面積A1+A2反映出來。從圖中可以看到,升高溫度,可以使活化分子百分數增大,從而使反應速率增大。
阿倫尼烏斯公式
非活化分子轉變為活化分子所需吸收的能量為活化能的計算可用阿倫尼烏斯方程求解。阿倫尼烏斯方程反應了化學反應速率常數K隨溫度變化的關係。在多數情況下,其定量規律可由阿倫尼烏斯公式來描述:
K=Aexp(-Ea/RT) (1)
式中:κ為反應的速率系(常)數;Ea和A分別稱為活化能和指前因子,是化學動力學中極重要的兩個參數;R為摩爾氣體常數;T為熱力學溫度。
(1)式還可以寫成:
lnκ=lnA-Ea/RT (2)
lnκ=與-1/T為直線關係,直線斜率為-Ea/R,截距為 lnA,由實驗測出不同溫度下的κ值,並將lnκ對1/T作圖,即可求出E值。
例:由Ea計算反應速率係數k
當已知某溫度下的k和Ea,可根據Arrhenius計算另一溫度下的k,或者與另一k相對應的溫度T。
2NO(g) = 2NO(g) + O(g)
已知:T=298.15K, k=0.469×10s
T=318.15K, k=6.29×10s 求:Ea及338.15K時的k。
Ea=[RTT(lnk/k)]/(T-T)=102kJ/mol
lnk/k=Ea[(1/T)-(1/T)]/R
K=6.12/1000S
對於更為複雜的描述κ與T的關係式中,活化能E定義為:
E=RT (dlnκ/dT)(3)
在元反應中,並不是反應物分子的每一次碰撞都能發生反應。S.A.阿倫尼烏斯認為,只有“活化分子”之間的碰撞才能發生反應,而活化分子的平均能量與反應物分子平均能量的差值即為活化能。近代反應速率理論進一步指出,兩個分子發生反應時必須經過一個過渡態——活化絡合物,過渡態具有比反應物分子和產物分子都要高的勢能,互撞的反應物分子必須具有較高的能量足以克服反應勢能壘,才能形成過渡態而發生反應,此即活化能的本質。
對於複合反應,由上述實驗方法求出的E值只是表觀值,沒有實際的物理意義。
阿侖尼烏斯(S.A.Arrhenius)發現化學反應的速度常數k和絕對溫度T之間有d(lnk)/dt=E/RT2的關係。這裡的E就是活化能。假若把上式積分得到lnk=lnA-(E/RT),從這個公式可知,在各種溫度下求得k值,把lnk對1/T作圖(這圖稱為阿侖尼烏斯圖)就得到直線,由於直線的斜率是-E/R,因而可求得E值。
活化能的物理意義一般認為是這樣:從原反應體繫到產物的中間階段存在一個過渡狀態,這個過渡狀態和原系統的能量差就是活化能E,而且熱能RT如不大於E,反應就不能進行。也就是原系統和生成物系統之間存在著能壘,其高度相當於活化能。其後埃林(H.Eyring)從過渡狀態(也叫做活性絡合物)和原系統之間存在著近似的平衡出發,對速度常數k導出了如下的關係:k=k(KT/h)exp(-ΔG*/RT)=k(KT/h)exp(ΔS*/R)exp(-ΔH*/RT)k為通透係數,K是波爾茲曼常數,h是普朗克常數,ΔG*、ΔS*、ΔH*分別為活化自由能、活化熵和活化焓。而且活化自由能與活化焓大致相等。酶促反應主要就是由於降低了活化自由能。
基本信息
- 中文名
- 活化能
- 外文名
- Activation energy
- 別名
- 閾能
- 定律
- 阿倫尼烏斯公式
- 學科
- 化學