校正曲線
校正曲線
校正曲線(calibration curve),用化學組成相同或結構相似的標準樣品,經過全分析過程所繪製的曲線。可用來對待測組分的輸入(如濃度)與響應輸出(如吸收強度)之間關係參量(如摩爾吸光係數)進行估計。校正曲線與標準曲線是不同的。
校正曲線是用於描述待測物質的濃度或量與相應的測量儀器的響應量或其他指示量之間的定量關係的曲線。校正曲線包括“工作曲線”和“標準曲線”。前者標準溶液的分析步驟與樣品分析步驟完全相同;後者標準溶液的分析步驟與樣品分析步驟相比有所省略,如省略樣品中的前處理。監測中常用校正曲線的直線部分。某一方法的標準曲線的直線部分所對應的待測物質濃度(或量)的變化範圍,稱為該方法的線性範圍。
建立校正曲線最常用的方法,是基於使偏差平方和達到極小的最小二乘法。從作圖的角度說,就是根據平面上一組離散的實驗點(x,y),(x,y),...,(x,y),選擇適當的連續曲線近似地擬合這一組離散實驗點,以儘可能完善地表示被測組分含量(或濃度)與檢測器響應值之間的關係。這種基於最小二乘原理研究因變數與自變數之間的相關關係的方法,稱為回歸分析。用回歸分析建立儀器分析校正曲線,因變數是儀器檢測器響應值,是具有概率分佈的隨機變數,自變數是被測組分含量(或濃度),為無概率分佈的固定變數。
校正曲線建立得好壞,直接影響測定結果的準確性。正確地建立校正曲線應遵循以下基本原則:
①從減小校正曲線的置信區間考慮,用4~6個實驗點建立校正曲線比較合適,實驗點要均勻分佈;
②在校正曲線的線性範圍內,要盡量擴大濃度的取值範圍,且被測組分含量要位於校正曲線中間;
③在總實驗工作量一定時,適當增加實驗點數目,減少實驗點重複測量次數是有利的;增加每個實驗點的重複測定次數只能提高個別實驗點的測定精密度,而增加實驗點數目能增加校正曲線的穩定性;
④在高濃度、低濃度兩端的濃度測定響應信號的精密度比校正曲線中央區域差,適當增加它們重複測量次數以提高它們的測定精密度;
⑤將空白溶液實驗點參與回歸,增加實驗點數目,以提高校正曲線的穩定性;按照校正曲線的標準偏差分
布曲線,在空白溶液(零濃度)時測定信號的標準偏差較大,因此不宜用空白溶液調零,應採用校正曲線截距扣空白,以減少零濃度點測定波動性對校正曲線定位的影響,提高空白扣除的準確度;
⑥校正曲線不得任意外延;
⑦當需要對校正曲線變動性進行校正時,宜用不同於製作工作曲線的濃度值重新標定校正曲線,以增加回歸線的實驗點數目,提高校正曲線的穩定性;
⑧不要用重新測定的個別實驗點,與原校正曲線的原點連接的直線(即重置斜率),或通過個別實驗點將曲線平移的方法來校正校正曲線的變動,最好將原實驗點與新測定的實驗點併合,重新建立校正曲線。
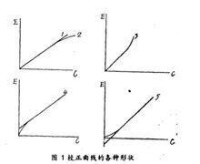
校正曲線
1、校正曲線不成直線的原因
影響校正曲線不成直線的原因是多方面的,主要為:
(1)單色光不夠純,使測得的吸光度有負誤差,溶液濃度越高,測定的負誤差越大,造成校正曲線上端向下彎曲(如圖1中曲線2)。
(2)在標準系列中,加入試劑的量不變而被測離子濃度不斷提高時,顯色劑過量的程度相對地減少,導致形成不同維成的絡合物,引起顏色強度的改變,校正曲線發生彎曲(如圖1中曲線2、3)。
(3)由於顯色產物是膠體溶液或被測離子的濃度高於一定限度時,因發生聚沉或締舍、離介等其他化學變化,使吸光度不隨濃度變化而成正比例改變,此時大多形成曲線2的校正曲線。
2.校正曲線不通過原點的原因
影響校正曲線不通過原點的原因大體上有以下幾方面;
(1)空白溶液的選擇和配製不當:
在測繪校正曲線的標準系列中,用空白溶液沒能恰好抵消干擾物質的吸光度,往往導致校正曲線平移而產生截距(如圖1中曲線4、5)。
(2)顯色反應和反應條件的問題:
當顯色反應的靈敏度不夠高時,被測物低於某一濃度就不能顯色;當顯色溶液中的掩蔽劑或緩衝溶液能絡合少量被測離子時,也有同樣現象,此時校正曲線成曲線5式,被測物質濃度低於濃度截距值時,.測得的吸光度都是零。
(3)比色皿的厚度或光學性能不一致:
盛空白溶液和盛顯色溶液的比色皿厚度或光學性能不一致時,都會使校正曲線不通過原點。如果盛顯色溶液的比色皿較薄或光的吸收或散射少些,校正曲線將呈“4”形狀。如果盛空白溶液的,比色皿或對光的吸收和散射多些,則呈“5”形狀。
應該指出,欲使校正曲線都成為通過原點的直線,有時不易做到。在實際工作中,最主要的是要求校正曲線重現性好,大體上成直線,就能滿足一般分析的要求。如果校正曲線經過採取措施仍不通過原點,若實踐證明其重現性好,則不通過原點也可以使用。