共找到16條詞條名為be的結果 展開
be
BE-生物等效性
生物等效性(bioequivalency,BE)是指一種藥物的不同製劑在相同實驗條件下,給予相同的劑量,其吸收速度與程度沒有明顯差別。
生物等效性(bioequivalency,BE)是指在同樣試驗條件下試驗製劑和對照標準製劑在藥物的吸收程度和速度的統計學差異。當吸收速度的差別沒有臨床意義時,某些藥物製劑其吸收程度相同而速度不同也可以認為生物等效。生物等效性與藥劑等效性(pharmaceuticalequivalents)不同,藥劑等效性是指同一藥物相同劑量製成同一劑型,但非活性成分不一定相同,在含量、純度、均勻度、崩解時間、溶出速率符合同一規定標準的製劑。藥劑等效性不能反映藥物製劑在體內情況。
1.改變劑型的產品。
2.改變處方與工藝的產品。
生物等效性用於評價兩個藥物對某疾病患者的效應(安全性和有效性)是否相同或相近。例如仿製葯與標準葯,天然葯與化學葯,口服藥與針劑,長效葯與短效葯,某葯低劑量與高劑量的比較需要用生物等效性方法來評價。FDA規定,若仿製藥品與註冊藥品間具有生物等效性,申報過程可按縮略申報程序(abbreviatednewdrugapplication,ANDA)進行,而不需要按新葯申報程序(newdrugapplication,NDA)進行,避免了耗時、昂貴的Ⅰ、Ⅲ期臨床試驗。所以,生物等效性檢驗在新葯臨床試驗中佔有極其重要的地位。
從等效性的程度來看有三種:平均生物等效性(averagebioequivalence,ABE),群體生物等效性(populationbioequivalence,PBE),個體生物等效性(individualbioequivalence,IBE)。
平均生物等效性:人群使用T藥物與使用R藥物所得效應,其平均值相同。ABE雖然保證平均效應相同,但不一定保證效應的變異度相同,即兩總體的均值相同,但方差不一定相同。
群體生物等效性:人群使用T藥物與使用R藥物所得效應,不僅其平均值相同,而且效應的變異度相同,即兩總體的邊緣分佈相同。PBE雖保證其邊緣分佈相同,但對每個個體而言,使用T藥物與使用R藥物所得效應不一定相同。即個體與藥物間可能存在交互作用(subject-byformulationinteraction)。
個體生物等效性:對每個個體而言,使用T藥物與使用R藥物所得效應值接近。
顯然,如果個體等效,則群體等效;如果群體等效,則平均等效,反之不然。從等效的程度來講,個體生物等效性最強,群體生物等效性其次,平均生物等效性最弱。從應用的角度來講,兩個具有個體生物等效性的藥物,具有用藥可交替性(switchability),即某患者在服用某藥物一段時間后,如果改用另一個與之具有個體等效性的藥物,可以得到同樣的效果;而具有群體生物等效性的藥物,具有處方可選擇性(prescribability),即醫生在給患者初次開藥時可以任意選用,這對於該類患者群體來說效應是相同的。可見三種等效性說明的問題不同,因此檢驗的方法不同,設計要求亦有別。
設定的背景
目前,國內外最常用的BE評價方法是葯動學方法,即採用生物利用度(Bioavailability,BA)指標進行BE評價。通常,BA指製劑中活性成分被吸收的程度和速度。用藥動學方法進行BE評價,就是考察藥學等效製劑或可替換藥品在相同試驗條件下,服用相同劑量,其活性成分吸收的程度和速度是否滿足預先設定的等效標準。在葯動學參數中,表徵吸收程度和速度的參數主要是AUC、Tmax和Cmax。因此,用藥動學方法評價製劑間是否具有生物等效性,就是以統計學方法評價試驗製劑與參比製劑測得的AUC、Tmax和Cmax等指標是否滿足預先設定的等效標準。預先設定的等效標準如何,也就成為影響BE評價的關鍵因素之一。
根據臨床醫生的建議以及FDA以往的經驗,對大多藥品來說,如果循環系統的藥物暴露差別在20%以內,將不會對臨床治療效果產生顯著影響。基於此點,FDA設定了試驗製劑和參比製劑的葯代動力學參數(AUC和Cmax)“差異應小於20%”作為等效性判定標準,具體判定方法為:通過雙單側t檢驗及(1-2α%)置信區間法,得到兩種製劑AUC或Cmax幾何均值比值的90%置信區間(ConfidenceInterval,CI),對於非窄治療窗的藥物,此90%CI必須落在80.00%~125.00%範圍內。另外,FDA和EMEA的指導原則還特彆強調,此置信區間必須保留兩位有效數字,並且不得通過四捨五入的方法,使受試藥物BE檢驗合格,即下限的最低值為80.00%,而上限不得超過125.00%,比如某項生物等效性試驗結果為79.96%~110.20%,則判定為生物不等效[2,3]。作為非正態分佈的Tmax,則要求用非參數的統計方法證明製劑間差異無統計學意義。
全球主要國家、組織和機構採用的生物等效性判定標準
同FDA要求一致,其他主要國家、地區的藥品監管機構(包括歐盟EMEA,日本厚生省)和世界衛生組織(WHO)都以80.00%~125.00%作為AUC和Cmax90%CI的等效性判定標準。在上述機構所制訂的指導原則中,對於AUC的等效性判定標準比較嚴格,通常只能縮小範圍(如:針對某些治療窗窄的藥物,EMEA建議可以縮小範圍至90.00%~111.11%)。[3,4]相對而言,Cmax的等效性判定標準具有一定的靈活性,比如加拿大藥品監管機構(HealthCanada)只要求Cmax均值的比值落在80~125%即可。[6]EMEA和WHO則提出,對於某些特殊情況的藥物(如高變異藥物,即葯動學參數的個體內差異在30%以上),可以根據情況適當擴大等效性判定標準的範圍,如EMEA建議對於個體內變異(CVintra)為35%的藥物,等效性判定標準可以擴大到77.23%~129.48%,當CVintra為40%時,該範圍可擴大至74.62%~134.02%,當CVintra為50%或以上則可以擴大至69.84%~143.19%。但申辦方必須提供證據證明,在此判定標準下,不會引起藥物安全性問題,並保證藥物的臨床療效沒有顯著差異,即需要證明Cmax差異的增大不會引起不良反應的顯著增加,也不會顯著影響療效。此外Cmax等效性判定標準範圍的擴大必須在BE試驗開始前設定,並提供相應的證據,而不能在試驗結束后,根據試驗結果更改。
日本厚生省則建議,如果擴大Cmax的等效性判定標準範圍,必須滿足以下三個條件:
(1)受試者人數不低於20,或在增加受試者人數之後總人數不低於30;
(2)Cmax均值的對數差值在log(0.9)~log(1.1)之間;
(3)對於體外溶出試驗,在任何的試驗條件下,當參比製劑體外溶出為30%,50%和80%時,受試製劑和參比製劑溶出度差別都在10%以內。[5]
我國目前的現行標準
我國2005年頒布的《化學藥物製劑人體生物利用度和生物等效性研究技術指導原則》中,AUC的90%CI的等效性判定標準和國際標準一致,而Cmax的標準,由於當時技術水平相對較低、臨床試驗條件等的限制,為方便BE試驗的管理和審評,統一設定了較為寬鬆的等效性判定標準,即70%~143%。[7]
近年來,隨著我國臨床葯動學試驗水平的進步和製劑研究水平的提升,對於藥品質量控制的要求將更加嚴格,以確保高質量仿製葯的開發。因此,參考先進國家與組織的規定,有必要提高Cmax的等效性判定標準要求,即採用80.00%-125.00%作為等效性判定標準。在此標準下,特殊藥物,如高變異藥物,可以適當擴大等效性判定標準範圍,但申辦者必須在BE試驗前提供相關安全性和臨床療效的證據,以及個體內變異情況的證據,在此基礎上重新設定等效性判定標準,如:75.00%-133.00%或者70.00%-143.00%。在試驗結束后,即使發現由於個體內差異很大,造成生物不等效,也不能根據結果再次對等效性判定標準的範圍進行放大。應當通過擴大受試者人數重新進行臨床試驗,降低標準偏差,來重新判定生物等效性。
色譜法:用於大多數藥物檢測
免疫法:多用於蛋白質多肽類物質檢測。
微生物學方法:可用於抗生素藥物的確定。
試驗設計與操作
交叉設計
受試者選擇
給藥劑量
取樣
參數計算。
標準化
數據處理和統計分析
數據表達
葯代動力學參數
統計分析
結果的評價
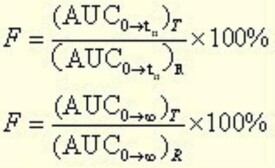
基本信息
- 中文名
- 生物等效性
- 外文名
- bioequivalency
- 定義
- 一種藥物的不同製劑在相同實驗條件下,給予相同的劑量,其吸收速度與程度沒有明顯差別
- 簡稱
- be
- 分析方法
- 色譜法、免疫法
- 類型
- 試驗方法