全基因組重測序
全基因組重測序
全基因組重測序是對已知基因組序列的物種進行不同個體的基因組測序,並在此基礎上對個體或群體進行差異性分析。SBC將不同梯度插入片段(Insert-Size)的測序文庫結合短序列(Short-Reads)、雙末端(Paired-End)進行測序,幫助客戶在全基因組水平上掃描並檢測與重要性狀相關的基因序列差異和結構變異,實現遺傳進化分析及重要性狀候選基因預測。
全基因組重測序是對已知基因組序列的物種進行不同個體的基因組測序,並在此基礎上對個體或群體進行差異性分析。全基因組重測序的個體,通過序列比對,可以找到大量的單核苷酸多態性位點(SNP),插入缺失位點(InDel,Insertion/Deletion)、結構變異位點(SV,Structure Variation)位點和拷貝數變異位點(CNV,copy number variation)。SBC可以協助客戶,通過生物信息手段,分析不同個體基因組間的結構差異,同時完成註釋。
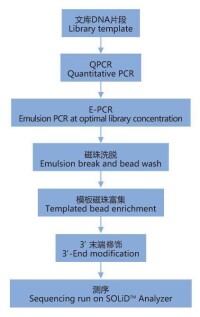
全基因組重測序
雙末端(Paired-End)測序原理
測序深度(Sequencing Depth):測序得到的鹼基總量(bp)與基因組大小(Genome)的比值,它是評價測序量的指標之一。測序深度與基因組覆蓋度之間是一個正相關的關係,測序帶來的錯誤率或假陽性結果會隨著測序深度的提升而下降。重測序的個體,如果採用的是Paired-End或Mate-Pair方案,當測序深度在10~15X以上時,基因組覆蓋度和測序錯誤率控制均得以保證。
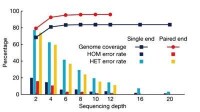
全基因組重測序
1.數據量產出
總鹼基數量、Totally mapped reads、Uniquely mapped reads統計,測序深度分析。
2.一致性序列組裝
與參考基因組序列(Reference genome sequence)的比對分析,利用貝葉斯統計模型檢測出每個鹼基位點的最大可能性基因型,並組裝出該個體基因組的一致序列。
3.SNP檢測及在基因組中的分佈
提取全基因組中所有多態性位點,結合質量值、測序深度、重複性等因素作進一步的過濾篩選,最終得到可信度高的SNP數據集。並根據參考基因組序列對檢測到的變異進行註釋。
4.InDel檢測及在基因組的分佈
在進行mapping的過程中,進行容Gap的比對並檢測可信的Short InDel。在檢測過程中,Gap的長度為1~5個鹼基。
5.Structure Variation檢測及在基因組中的分佈
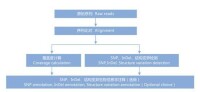
全基因組重測序
基本信息
- 中文名
- 全基因組重測序