共找到2條詞條名為假性肥大型肌營養不良的結果 展開
- 假肥大型肌營養不良症
- 假肥大性肌營養不良
假性肥大型肌營養不良
假肥大性肌營養不良
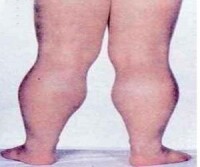
假肥大型肌營養不良包括杜興肌營養不良(DMD)和貝克肌營養不良(BMD),臨床上常見的是前一種,是原發於肌肉組織的X連鎖隱性遺傳的肌病。本病為遺傳性疾病,多屬X連鎖隱性遺傳,個別為染色體隱性遺傳。DMD與BMD是等位基因病。Gowers征:本病的患兒由仰卧位坐起時,有一個特徵性的起立動作,即患兒不能直接從仰卧位上坐起,需首先翻身成為俯卧位,然後再蹲起,再轉換為四點支持位。腓腸肌假性肥大:絕大多數的患兒有腓腸肌的假性肥大,可見雙側的腓腸肌肥大,這是由於萎縮的肌纖維組織被脂肪充填而致,同時出現肌力減弱,但觸之堅硬。
本病的發生是由於編碼dystrophin的DMD基因的突變所引起,有約 1/3 病例為散發,沒有家族史,是由基因突變造成。 Becker 肌營養不良 (Becker muscular dystrophy, BMD) 的產生也是由於 DMD 基因突變所引起,通常突變后產生的異常 DMD 蛋白仍具有一定功能,因而臨床癥狀較 DMD 輕得多。
患兒出生時的活動如抬頭、坐姿等均正常,自 1 歲以後開始逐漸出現站立和行走困難,首先影響骨盆帶肌肉,以後累及肩胛帶肌肉。患兒動作笨拙,易跌倒,走路搖搖晃晃,登樓梯或由坐、卧位起立困難。隨著病變的進展,臀中肌無力導致行走時呈特殊的鴨步,患兒從仰卧位起立時需先翻轉為俯卧,再以雙手支持地面和下肢緩慢地站立,稱為 Gower 氏征。患兒雙側腓腸肌逐漸呈假性肥大,腱反射減弱或消失。部分患者表現為行為異常。病變呈進行性加重,常到 10 歲時已不能行走,大多數患兒最終卧床不起,併發痙攣、褥瘡、肺炎而在 20 歲前死亡。 BMD (良性假肥大性肌營養不良症)患者的臨床癥狀一般較輕,可存活到成年以後。
約 1/4 的患兒有智力低下,血中磷酸肌酸激酶( CRK )濃度顯著升高,可達正常值的 1萬倍,肌電圖顯示肌病的改變伴有較輕的失神經支配電位;肌肉活體組織檢查可見肌纖維壞死與再生同時存在,並有結締組織增生。
根據患者特有的癥狀和體征,結合血 CPK 酶學檢查和肌電圖檢查結果,一般不難作出診斷。確診可根據多重 PCR 技術、 Southern 雜交、點突變檢查等方法。對於突變未明確的家系,可用 STR 位點進行連鎖分析,用於攜帶者的檢出或產前診斷。
目前對於 DMD 尚無有效療法,唯一有效的預防途徑是對高風險胎兒進行產前診斷,確診后流產。
本病屬 X 連鎖隱性遺傳病。致死性散發性 X -連鎖隱性遺傳的 1/3 病例都是由新發生的基因突變引起 ( 又稱 Haldane 規律 ) ,這在家系分析時應特別注意。
常見諮詢問題:
1.家族中沒有發現類似病例,是否也有可能患 DMD?
前面已經提到, DMD中,有約1/3病例是由新發生的基因突變引起,即父母雙親中,DMD基因都正常,但個體胚胎髮育時,出現了DMD基因的突變,導致疾病的發生。這種突變發生的個體,可將致病基因遺傳給後代。
2.如何通過性別選擇,規避 DMD的遺傳風險?
本病主要為男性發病,在已生育過 DMD患者的家庭中,可通過 產前診斷,選擇女性胎兒,降低DMD的發生風險。如果能應用基因診斷方法,對所懷男胎排除DMD基因的突變,仍可生育正常男胎。
假肥大性肌營養不良