線粒體基因病
生物學術語
線粒體基因病(Mitochondrial genic disorders)線粒體基因組中發生基因突變所導致的一類疾病,其傳遞和表達完全不同於由核基因突變引起的疾病,是一組獨特的遺傳病,稱為線粒體基因病。
目錄
就目前所知,線粒體基因病是由於線粒體DNA發生了重複、缺失或點突變,呈母系遺傳,父源性線粒體傳遞只是散發性的偶然事件。據對眼肌麻痹、視網膜變性及心肌綜合症這些線粒體基因病家系的調查顯示,其中51個母親(94%)傳遞了此症,但傳遞此症的父親只有3個(6%)。此外,線粒體DNA基因突變的傳遞有一定數量上的特點。每個細胞中細胞質內所含有的線粒體分子甚多,如果細胞內所有這些線粒體DNA分子上的某一基因座都是同一基因,即同為正常基因或同為突變基因,則該細胞為純質的;如果一個細胞的所有線粒體DNA在同一基因座上同時存在正常基因和突變基因,則該細胞為異質的。
線粒體基因病的特點:
1、母系遺傳(matrilinear inheritance)
卵子與精子細胞核的結合是對等的,但細胞質的結合是遠遠不對等的。在絕大多數情況下,突變的線粒
體DNA通過母親卵子細胞質的線粒體傳給子代,通過父親傳遞的極為罕見。
2、數量概念
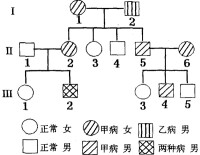
線粒體基因病系譜圖
但如一個細胞的數千個mtDNA分子在這個位點上同時存在正常基因和突變基因,這就成為雜質(heteroplasmy) 。
(1)一般說,突變的mtDNA的數量超過一定限度時,會出現臨床癥狀。(閾值)
(2)突變mtDNA所佔比例似與臨床癥狀的表現程度相關。
3、傳遞突變的母親可為患者,也可是表現正常的雜質攜帶者。
線粒體基因病病例:
1、Leber遺傳性視神經病(Leber hereditary optic neuropathy,LHON)是這類疾病的典型例子之一。這種疾病是由於線粒體呼吸鏈複合物遺傳性異常而引起的。病初發時為急性或亞急性眼球的神經炎,引發嚴重雙側視神經萎縮和大片中心暗點,使視力突然喪失並伴有色覺障礙。在急性發作之後,視覺進一步衰退,但常可維持0.02~0.5的視力。此症發病高峰年齡是20~25歲,但任何年齡段都可能發病。
臨床癥狀:通常在成年期發病(20-25歲)
(1)急性或亞急性眼球后神經炎
(2)急性發作后,視覺逐步衰退
(3)色覺障礙
(4)其他神經異常,如智力障礙
基因突變:遺傳異質性
涉及呼吸鏈複合物I NADH-普醌氧化還原酶第1亞單位肽鏈的基因:
MTND1*LHON4160T→C,約佔50-75%(亮→脯)
MTND1*LHON3460G→A,約佔15-25%(丙→絲)
MTND1*LHON3394T→C,約佔15-25%(酪→組)
2、線粒體腦肌病、乳酸酸中毒及卒中樣發作綜合征(mitochondrial encephalomyopathy, lacticacidosis,and stroke-like episodes,MELAS)
臨床癥狀:
(1)粗糙紅纖維
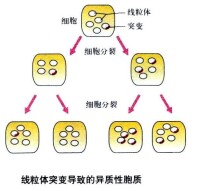
線粒體基因異質性傳遞系譜圖
(3)乳酸酸中毒
(4)感覺神經性聽覺喪失
(5)痴獃
(6)反覆發生卒中樣發作,頭痛、嘔吐
基因突變:遺傳異質性
線粒體tRNALeu基因突變:
MTTL1*MELAS 3243 T →C或A →G
呼吸鏈複合物I NADH-普醌氧化還原酶第4亞單位肽鏈的基因突變:
MTND4*MELAS 11084A →G
3、人類神經性肌肉衰弱症也是線粒體基因控制的遺傳病。

基本信息
- 中文名
- 線粒體基因病