高效液相色譜
又稱“高壓液相色譜”、“高速液相色譜”等
高效液相色譜法(High Performance Liquid Chromatography\HPLC)又稱“高壓液相色譜”、“高速液相色譜”、“高分離度液相色譜”、“近代柱色譜”等。高效液相色譜是色譜法的一個重要分支,以液體為流動相,採用高壓輸液系統,將具有不同極性的單一溶劑或不同比例的混合溶劑、緩衝液等流動相泵入裝有固定相的色譜柱,在柱內各成分被分離后,進入檢測器進行檢測,從而實現對試樣的分析。該方法已成為化學、醫學、工業、農學、商檢和法檢等學科領域中重要的分離分析技術應用。
1903年俄國植物化學家茨維特(Tswett)首次提出“色譜法”(Chromatography)和“色譜圖”(Chromatogram)的概念。茨維特使用色譜法chromatography(來自希臘字,chroma意思是顏色,graphy意思是記錄-直譯為顏色記錄)來描述他的彩色試驗。(令人好奇的是,俄羅斯名字茨維特意思是顏色。)他在論文中寫到:
“(原文)一植物色素的石油醚溶液從一根主要裝有碳酸鈣吸附劑的玻璃管上端加入,沿管濾下,後用純石油醚淋洗,結果按照不同色素的吸附順序在管內觀察到它們相應的色帶,就象光譜一樣,稱之為色譜圖。”
1930年以後,相繼出現了紙色譜、離子交換色譜和薄層色譜等液相色譜技術。
1952年,英國學者Martin和Synge基於他們在分配色譜方面的研究工作,提出了關於氣-液分配色譜的比較完整的理論和方法,把色譜技術向前推進了一大步,這是氣相色譜在此後的十多年間發展十分迅速的原因。
1958年,基於Moore和Stein的工作,離子交換色譜的儀器化導致了氨基酸分析儀的出現,這是近代液相色譜的一個重要嘗試,但分離效率尚不理想。
1960年中後期,氣相色譜理論和實踐發展,以及機械、光學、電子等技術上的進步,液相色譜又開始活躍。到60年代末期把高壓泵和化學鍵合固定相用於液相色譜就出現了HPLC。
1970年中期以後,微處理機技術用於液相色譜,進一步提高了儀器的自動化水平和分析精度。
1990年以後,生物工程和生命科學在國際和國內的迅速發展,為高效液相色譜技術提出了更多、更新的分離、純化、製備的課題,如人類基因組計劃,蛋白質組學有HPLC作預分離等。
高效液相色譜法有“四高一廣”的特點:
①高壓:流動相為液體,流經色譜柱時,受到的阻力較大,為了能迅速通過色譜柱,必須對載液加高壓。
②高速:分析速度快、載液流速快,較經典液體色譜法速度快得多,通常分析一個樣品在15~30分鐘,有些樣品甚至在5分鐘內即可完成,一般小於1小時。
③高效:分離效能高。可選擇固定相和流動相以達到最佳分離效果,比工業精餾塔和氣相色譜的分離效能高出許多倍。
④高靈敏度:紫外檢測器可達0.01ng,進樣量在μL數量級。
⑤應用範圍廣:百分之七十以上的有機化合物可用高效液相色譜分析,特別是高沸點、大分子、強極性、熱穩定性差化合物的分離分析,顯示出優勢。
⑥柱子可反覆使用:用一根柱子可分離不同化合物
⑦樣品量少、容易回收:樣品經過色譜柱后不被破壞,可以收集單一組分或做製備。
此外高效液相色譜還有色譜柱可反覆使用、樣品不被破壞、易回收等優點,但也有缺點,與氣相色譜相比各有所長,相互補充。高效液相色譜的缺點是有“柱外效應”。在從進樣到檢測器之間,除了柱子以外的任何死空間(進樣器、柱接頭、連接管和檢測池等)中,如果流動相的流型有變化,被分離物質的任何擴散和滯留都會顯著地導致色譜峰的加寬,柱效率降低。高效液相色譜檢測器的靈敏度不及氣相色譜。
1.吸附色譜法(AdsorptionChromatography)
2.分配色譜法(PartitionChromatography)
3.離子色譜法(IonChromatography)
4.分子排阻色譜法/凝膠色譜法(SizeExclusionChromatography)
5.鍵合相色譜法(bonded-phasechromatography)
6.親和色譜法(AffinityChromatography)
可分為“高壓輸液泵”、“色譜柱”、“進樣器”、“檢測器”、“餾分收集器”以及“數據獲取與處理系統”等部分。
液相色譜和質譜連接,可以增加額外的分析能力,能夠準確鑒定和定量像細胞和組織裂解液,血液,血漿,尿液和口腔液等複雜樣品基質中的微量化合物。高效液相色譜質譜系統(ABSciexEksigentLC/MS和LC/MS/MS)提供了一些獨特的優勢,包括:
● 快速分析和流轉所需的最少樣品準備
● 高靈敏度並結合可分析多個化合物能力,甚至可以跨越化合物的種類
● 高精確度,高解析度鑒定和量化目標分析物
功能
驅動流動相和樣品通過色譜分離柱和檢測系統;
性能要求
流量穩定(±1),耐高壓(30~60Mpa),耐各種流動相:例如:有機溶劑、水和緩衝液;
種類
往複泵和隔膜泵。
功能
分離樣品中的各個物質;
尺寸
10~30cm長,2~5mm內徑的內壁拋光的不鏽鋼管柱;
填料粒度
5~10μm,高效微粒固定相;
功能
將待分析樣品引入色譜系統;
種類
①注射器,10Mpa以下,1~10μm微量注射器進樣
②停流進樣
③閥進樣,常用、較理想、體積可變,可固定
④自動進樣器,有利於重複操作,實現自動化
檢測器
功能
將被分析組在柱流出液中濃度的變化轉化為光學或電學信號;
分類
①示差折光化學檢測器
②紫外吸收檢測器
③紫外-可見分光光度檢測器
④二極體陣列紫外檢測器
⑤熒光檢測器
⑥電化學檢測器
功能
如果所進行的色譜分離不是為了純粹的色譜分析,而是為了做其它波譜鑒定,或獲取少量試驗樣品的小型製備,餾分收集是必要的;
方法
①手工,少數幾個餾分,手續麻煩,易出差錯。
②餾分收集器收集,比較理想,微機控制操作準確。
獲取與處理功能
把檢測器檢測到的信號顯示出來。
(Liquid-liquidPartitionChromatography)及化學鍵合相色譜(ChemicallyBondedPhaseChromatography) 流動相和固定相都是液體。流動相與固定相之間應互不相溶(極性不同,避免固定液流失),有一個明顯的分界面。當試樣進入色譜柱,溶質在兩相間進行分配。達到平衡時,服從於高效液相色譜計算公式:
式中,cs—溶質在固定相中濃度;cm—溶質在流動相中的濃度;Vs—固定相的體積;Vm—流動相的體積。LLPC與GPC有相似之處,即分離的順序取決於K,K大的組分保留值大;但也有不同之處,GPC中,流動相對K影響不大,LLPC流動相對K影響較大。
a.正相液-液分配色譜法(NormalPhaseliquidChromatography):流動相的極性小於固定液的極性。
b.反相液-液分配色譜法(ReversePhaseliquidChromatography):流動相的極性大於固定液的極性。
c.液-液分配色譜法的缺點:儘管流動相與固定相的極性要求完全不同,但固定液在流動相中仍有微量溶解;流動相通過色譜柱時的機械衝擊力,會造成固定液流失。上世紀70年代末發展的化學鍵合固定相(見后),可克服上述缺點。
流動相為液體,固定相為吸附劑(如硅膠、氧化鋁等)。這是根據物質吸附作用的不同來進行分離的。其作用機制是:當試樣進入色譜柱時,溶質分子(X)和溶劑分子(S)對吸附劑表面活性中心發生競爭吸附(未進樣時,所有的吸附劑活性中心吸附的是S),可表示如下:XmnSa======XanSm
式中:Xm--流動相中的溶質分子;Sa--固定相中的溶劑分子;Xa--固定相中的溶質分子;Sm--流動相中的溶劑分子。
當吸附競爭反應達平衡時:
K=[Xa][Sm]/[Xm][Sa]
式中:K為吸附平衡常數。[討論:K越大,保留值越大。]
(Ion-exchangeChromatography)
IEC是以離子交換劑作為固定相。IEC是基於離子交換樹脂上可電離的離子與流
動相中具有相同電荷的溶質離子進行可逆交換,依據這些離子以交換劑具有不同的親和力而將它們分離。以陰離子交換劑為例,其交換過程可表示如下:
X-(溶劑中)(樹脂-R4NCl-)===(樹脂-R4NX-)Cl-(溶劑中)
當交換達平衡時:
KX=[-R4NX-][Cl-]/[-R4NCl-][X-]
分配係數為:
DX=[-R4NX-]/[X-]=KX[-R4NCl-]/[Cl-]
[討論:DX與保留值的關係]
凡是在溶劑中能夠電離的物質通常都可以用離子交換色譜法來進行分離。
(IonPairChromatography)
離子對色譜法是將一種(或多種)與溶質分子電荷相反的離子(稱為對離子或反離子)加到流動相或固定相中,使其與溶質離子結合形成疏水型離子對化合物,從而控制溶質離子的保留行為。其原
理可用下式表示:X水相Y-水相===XY-有機相
式中:X水相--流動相中待分離的有機離子(也可是陽離子);Y-水相--流動相中帶相反電荷的離子對(如氫氧化四丁基銨、氫氧化十六烷基三甲銨等);XY---形成的離子對化合物。
當達平衡時:
KXY=[XY-]有機相/[X]水相[Y-]水相
根據定義,分配係數為:
DX=[XY-]有機相/[X]水相=KXY[Y-]水相
[討論:DX與保留值的關係]
離子對色譜法(特別是反相)解決了以往難以分離的混合物的分離問題,諸如酸、鹼和離子、非離子混合物,特別是一些生化試樣如核酸、核苷、生物鹼以及藥物等分離。
(IonChromatography)
用離子交換樹脂為固定相,電解質溶液為流動相。以電導檢測器為通用檢測器,為消除流動相中強電解質背景離子對電導檢測器的干擾,設置了抑制柱。試樣組分在分離柱和抑制柱上的反應原理與離子交換色譜法相同。
以陰離子交換樹脂(R-OH)作固定相,分離陰離子(如Br-)為例。當待測陰離子Br-隨流動相(NaOH)進入色譜柱時,發生如下交換反應(洗脫反應為交換反應的逆過程):
抑制柱上發生的反應:
R-HNaOH-===R-NaH2O
R-HNaBr-===R-NaHBr-
可見,通過抑制柱將洗脫液轉變成了電導值很小的水,消除了本底電導的影響;試樣陰離子Br-則被轉化成了相應的酸HBr-,可用電導法靈敏的檢測。
離子色譜法是溶液中陰離子分析的最佳方法。也可用於陽離子分析。
(StericExclusionChromatography)
空間排阻色譜法以凝膠(gel)為固定相。它類似於分子篩的作用,但凝膠的孔徑比分子篩要大得多,一般為數納米到數百納米。溶質在兩相之間不是靠其相互作用力的不同來進行分離,而是按分子大小進行分離。分離只與凝膠的孔徑分佈和溶質的流動力學體積或分子大小有關。試樣進入色譜柱后,隨流動相在凝膠外部間隙以及孔穴旁流過。在試樣中一些太大的分子不能進入膠孔而受到排阻,因此就直接通過柱子,首先在色譜圖上出現,一些很小的分子可以進入所有膠孔並滲透到顆粒中,這些組分在柱上的保留值最大,在色譜圖上最後出現。
流程:如右圖所示,溶劑貯器⑴中的流動相被泵⑵吸入,經〔3〕梯度控制器按一定的梯度進行混合然後輸出,經⑷測其壓力和流量,導入(5進樣閥(器)經⑹保護柱、⑺分離柱後到⑻檢測器檢測,由⑽數據處理設備處理數據或⑾記錄儀記錄色譜圖,⑿餾分收集器收集餾分,⒀為廢液。
色譜圖(chromatogram)——樣品流經色譜柱和檢測器,所得到的信號-時間曲線,又稱色譜流出曲線(elutionprofile)。
基線(baseline)——經流動相衝洗,柱與流動相達到平衡后,檢測器測出一段時間的流出曲線。一般應平行於時間軸。
噪音(noise)——基線信號的波動。通常因電源接觸不良或瞬時過載、檢測器不穩定、流動相含有氣泡或色譜柱被污染所致。
漂移(drift)——基線隨時間的緩緩變化。主要由於操作條件如電壓、溫度、流動相及流量的不穩定所引起,柱內的污染物或固定相不斷被洗脫下來也會產生漂移。
色譜峰(peak)——組分流經檢測器時響應的連續信號產生的曲線。流出曲線上的突起部分。正常色譜峰近似於對稱形正態分佈曲線(高斯Gauss曲線)。不對稱色譜峰有兩種:
前延峰(leadingpeak)和拖尾峰(tailingpeak)。前者少見。
峰底—基線上峰的起點至終點的距離。
峰高(peakheight,h)—峰的最高點至峰底的距離。
峰寬(peakwidth,W)—峰兩側拐點處所作兩條切線與基線的兩個交點間的距離。W=4σ
半峰寬(peakwidthathalf-height,Wh/2)—峰高一半處的峰寬。Wh/2=2.355σ
峰面積(peakarea,A)—峰與峰底所包圍的面積。
保留時間(retentiontime,tR)——從進樣開始到某個組分在柱后出現濃度極大值的時間。
理論塔板數(theoreticalplatenumber,N)——用於定量表示色譜柱的分離效率(簡稱柱效)。
分離度(resolution,R)——相鄰兩峰的保留時間之差與平均峰寬的比值。也叫解析度,表示相鄰兩峰的分離程度。R≥1.5稱為完全分離。
《中國藥典》規定R應大於1.5。
拖尾因子(tailingfactor,T)——T=,用以衡量色譜峰的對稱性。也稱為對稱因子(symmetryfactor)或不對稱因子(asymmetryfactor)。
《中國藥典》規定T應為0.95~1.05。
T1.05為拖尾峰。
填料和流動相的組分
應按各品種項下的規定,常用的色譜柱填料有硅膠和化學鍵合硅膠。後者以十八烷基硅烷鍵合硅膠最為常用辛基鍵合硅膠次之,氰基或氨基鍵合硅膠也有使用;離子交換填料用於離子交換色譜;凝膠或玻璃微球等,用於分子排阻色譜等。注樣量一般為數微升。除另有規定外,柱溫為室溫,檢測器為紫外吸收檢測器。
在用紫外吸收檢測器時,所用流動相應符合紫外分光光度法項下對溶劑的要求。
正文中各品種項下規定的條件除固定相種類、流動相組分、檢測器類型不得任意改變外,其餘如色譜柱內徑、長度、固定相牌號、載體粒度、流動相流速、混合流動相各組分的比例、柱溫、進樣量、檢測器的靈敏度等,均可適當改變。
以適應具體品種並達到系統適用性試驗的要求。一般色譜圖約於20分鐘內記錄完畢。
系統適用性
按各品種項下要求對儀器進行適用性試驗,即用規定的對照品對儀器進行試驗和調整,應達到規定的要求;或規定分析狀態下色譜柱的最小理論板數、分離度和拖尾因子.
色譜柱
在選定的條件下,注入供試品溶液或各品種項下規定的內標物質溶液,記錄色譜圖,量出供試品主成分或內標物質峰的保留時間t(R)和半高峰寬W(h/2),按n=5.54[t(R)╱W(h/2)]^2計算色譜柱的理論板數,如果測得理論板數低於各品種項下規定的最小理論板數,應改變色譜柱的某些條件(如柱長、載體性能、色譜柱充填的優劣等),使理論板數達到要求。
分離度
定量分析時,為便於準確測量,要求定量峰與其他峰或內標峰之間有較好的分離度。分離度(R)的計算公式為:R=2(tR2-tR1)/(W1+W2),式中t(R2)為相鄰兩峰中后一峰的保留時間;t(R1)為相鄰兩峰中前一峰的保留時間;W1及W2為此相鄰兩峰的峰寬。除另外有規定外,分離度應大於1.5。
拖尾因子
為保證測量精度,特別當採用峰高法測量時,應檢查待測峰的拖尾因子(T)是否符合各品種項下的規定或不同濃度進樣的校正因子誤差是否符合要求。拖尾因子計算公式為:
T(拖尾因子)=W0.05h/2d1式中W(0.05h)為0.05峰高處的峰寬;
d1為峰極大至峰前沿之間的距離。除另有規定外,T應在0.95~1.05間。
也可按各品種校正因子測定項下,配製相當於80%、100%和120%的對照品溶液,加入規定量的內標溶液,配成三種不同濃度的溶液,分別注樣3次,計算平均校正因子,其相對標準偏差應不大於2.0%。
定量測定時,可根據樣品的具體情況採用峰面積法或峰高法。但用歸一法或內標法測定雜質總量時,須採用峰面積法。
面積歸一化法
測定供試品(或經衍生化處理的供試品)中各雜質及雜質的總量限度採用不加校正因子的峰面積歸一法。計算各雜質峰面積及其總和,並求出佔總峰面積的百分率。但溶劑峰不計算在內。色譜圖的記錄時間應根據各品種所含雜質的保留時間決定,除另有規定外可為該品種項下主成分保留時間的倍數。
主成分自身對照法
當雜質峰面積與成分峰面積相差懸殊時,採用主成分自身對照法。在測定前,先按各品種項下規定的雜質限度,將供試品稀釋成一定濃度的溶液作為對照溶液,進樣,調節檢測器的靈敏度或進樣量,使對照溶液中的主成分色譜峰面積滿足準確測量要求。然後取供試品溶液,進樣,記錄時間,除另有規定外,應為主成分保留時間的倍數。根據測得的供試品溶液的各雜質峰面積及其總和並和對照溶液主成分的峰面積比較,計算雜質限度。
內標法 測定供試品中雜質的總量限度
採用不加校正因子的峰面積法。取供試品,按各品種項下規定的方法配製不含內標物質的供試品溶液,注入儀器,記錄色譜圖Ⅰ;再配製含有內標物質的供試品溶液,在同樣的條件下注樣,記錄色譜圖Ⅱ。記錄的時間除另有規定外,應為該品種項下規定的內標峰保留時間的倍數,色譜圖上內標峰高應為記錄儀滿標度的30%以上,否則應調整注樣量或檢測器靈敏度。
如果色譜圖Ⅰ中沒有與色譜圖Ⅱ上內標峰保留時間相同的雜質峰,則色譜圖Ⅱ中各雜質峰面積之和應小於內標物質峰面積(溶劑峰不計在內)。如果色譜圖Ⅰ中有與色譜圖Ⅱ上內標物質峰保留時間相同的雜質峰,應將色譜圖Ⅱ上的內標物質峰面積減去色譜圖Ⅰ中此雜質峰面積,即為內標物質峰的校正面積;色譜圖Ⅱ中各雜質峰總面積加色譜圖Ⅰ中此雜峰面積,即為各雜質峰的校正總面積,各雜質峰的校正總面積應小於內標物質峰的校正面積。
加校正因子測定供試品主成分含量
按各品種項下的規定,精密稱(量)取對照品和內標物質,分別配成溶液,精密量取各溶液,配成校正因子測定用的對照溶液,取一定量注入儀器,記錄色譜圖,測量對照品和內標物質的峰面積或峰高,按下式計算校正因子:
As/ms]校正因子f=-Ar/mr式中As為內標物質的峰面積或峰高,Ar為對照品的峰面積或峰高;ms為加入內標物質的量mr為加入對照品的量。再取各品種項下含有內標物質的供試品溶液,注入儀器,記錄色譜圖,測量供試品(或其雜質)峰和內標物質的峰面積或峰高,按下式計算含量:Ax含量(mx)=f×-As/ms式中Ax為供試品(或其雜質)峰面積或峰高;mx為供試品(或其雜質)的量。f、As和ms的意義同上。
當配製校正因子測定用的對照溶液和含有內標物質的供試品溶液使用同一分內標物質溶液時,則配製內標物質溶液不必精密稱(量)取。
外標法 測定供試品中含量
按各品種項下的規定,精密稱(量)取對照品和供試品,配製成溶液,分別精密取一定量,注入儀器,記錄色譜圖,測量對照品和供試品待測成分的峰面積(或峰高),按下式計算含量:
A<[x]>含量(mx)=mr×-Ar式中各符號意義同上
由於微量注射器不易精確控制進樣量,當採用外標法測定供試品中某雜質或主成分含量時,以定量環進樣為好。
要正確地選擇色譜分離方法,首先必須儘可能多地了解樣品的有關性質,其次必須熟悉各種色譜方法的主要特點及其應用範圍。選擇色譜分離方法的主要根據是樣品的相對分子質量的大小,在水中和有機溶劑中的溶解度,極性和穩定程度以及化學結構等物理、化學性質。
對於相對分子質量較低(一般在200以下),揮發性比較好,加熱又不易分解的樣品,可以選擇氣相色譜法進行分析。相對分子質量在200~2000的化合物,可用液固吸附、液-液分配和離子交換色譜法。相對分子質量高於2000,則可用空間排阻色譜法。
水溶性樣品最好用離子交換色譜法和液液分配色譜法;微溶於水,但在酸或鹼存在下能很好電離的化合物,也可用離子交換色譜法;油溶性樣品或相對非極性的混合物,可用液-固色譜法。
若樣品中包含離子型或可離子化的化合物,或者能與離子型化合物相互作用的化合物(例如配位體及有機螯合劑),可首先考慮用離子交換色譜,但空間排阻和液液分配色譜也都能順利地應用於離子化合物;異構體的分離可用液固色譜法;具有不同官能團的化合物、同系物可用液液分配色譜法;對於高分子聚合物,可用空間排阻色譜法。
HPLC的出現不過三十多年的時間,但這種分離分析技術的發展十分迅猛,應用十分廣泛。其儀器結構和流程多種多樣。典型的高效液相色譜儀結構和流程可用下列方框圖表示(SeeFig.3-4)。高效液相色譜儀一般都具備貯液器、高壓泵、梯度洗提裝置(用雙泵)、進樣器、色譜柱、檢測器、恆溫器、記錄儀等主要部件。
高效液相色譜更適宜於分離、分析高沸點、熱穩定性差、有生理活性及相對分子量比較大的物質,因而廣泛應用於核酸、肽類、內酯、稠環芳烴、高聚物、藥物、人體代謝產物、表面活性劑,抗氧化劑、殺蟲劑、除莠劑的分析等物質的分析。
HPLC使用的色譜柱是很細的(1~6mm),所用固定相的粒度也非常小(幾μm到幾十μm),所以流動相在柱中流動受到的阻力很大,在常壓下,流動相流速十分緩慢,柱效低且費時。為了達到快速、高效分離,必須給流動相施加很大的壓力,以加快其在柱中的流動速度。為此,須用高壓泵進行高壓輸液。高壓、高速是高效液相色譜的特點之一。HPLC使用的高壓泵應滿足下列條件:
a.流量恆定,無脈動,並有較大的調節範圍(一般為1~10mL/min);
b.能抗溶劑腐蝕;
c.有較高的輸液壓力;對一般分離,60×10^5Pa的壓力就滿足了,對高效分離,要求達到150~300×10^5Pa。
⑴往複式柱塞泵
當柱塞推入缸體時,泵頭出口(上部)的單向閥打開,同時,流動相進入的單向閥(下部)關閉,這時就輸出少量的流體。反之,當柱塞向外拉時,流動相入口的單向閥打開,出口的單向閥同時關閉,一定量的流動相就由其儲液器吸入缸體中。這種泵的特點是不受整個色譜體系中其餘部分阻力稍有變化的影響,連續供給恆定體積的流動相。
⑵氣動放大泵
其工作原理是:壓力為p1的低壓氣體推動大面積(SA)活塞A,則在小面積(SB)活塞B輸出壓力增大至p2的液體。壓力增大的倍數取決於A和B兩活塞的面積比,如果A與B的面積之比為50:1,則壓力為5×Pa的氣體就可得到壓力為250×Pa的輸出液體。這是一種恆壓泵。
類似於GC中的程序升溫。已成為現代高效液相色譜中不可缺少的部分。梯度洗提,就是載液中含有兩種(或更多)不同極性的溶劑,在分離過程中按一定的程序連續改變載液中溶劑的配比和極性,通過載液中極性的變化來改變被分離組分的分離因素,以提高分離效果。梯度洗提可以分為兩種:
a.低壓梯度(也叫外梯度):在常壓下,預先按一定程序將兩種或多種不同極性的溶劑混合后,再用一台高壓泵輸入色譜柱。
b.高壓梯度(或稱內梯度系統):利用兩台高壓輸液泵,將兩種不同極性的溶劑按設定的比例送入梯度混合室,混合后,進入色譜柱。
⑴注射器進樣裝置:進樣所用微量注射器及進樣方式與GC法一樣。進樣壓力150×10^5Pa時,必須採用停流進樣。⑵高壓定量進樣閥:與GC法用的流通法相似,能在高壓下進樣。
色譜柱是色譜儀最重要的部件(心臟)。通常用厚壁玻璃管或內壁拋光的不鏽鋼管製作的,對於一些有腐蝕性的樣品且要求耐高壓時,可用銅管、鋁管或聚四氟乙烯管。柱子內徑一般為1~6mm。常用的標準柱型是內徑為4.6或3.9mm,長度為15~30cm的直形不鏽鋼柱。填料顆粒度5~10μm,柱效以理論塔板數計大約7000~10000。
發展趨勢是減小填料粒度和柱徑以提高柱效。
⑴紫外光度檢測器
它的作用原理是基於被分析試樣組分對特定波長紫外光的選擇性吸收,組分濃度與吸光度的關係遵守比爾定律。最常用的檢測器,應用最廣,對大部分有機化合物有響應。
特點:
a.靈敏度高:其最小檢測量10-9g·mL-1,故即使對紫外光吸收很弱的物質,也可以檢測;
b.線性範圍寬;(比爾定律)
c.流通池可做得很小(1mm×10mm,容積8μL);
d.對流動相的流速和溫度變化不敏感可用於梯度洗脫;
e.波長可選,易於操作:如,使用裝有流通池的可見紫外分光光度計(可變波長檢測器)。
缺點:對紫外光完全不吸收的試樣不能檢測;同時溶劑的選擇受到限制。
⑵光電二極體陣列檢測器
紫外檢測器的重要進展;陣列由1024個光電二極體陣列,每個光電二極體寬僅50μm,各檢測一窄段波長。如圖所示,在檢測器中,光源發出的紫外或可見光通過液相色譜流通池,在此流動相中的各個組分進行特徵吸收,然後通過狹縫,進入單色其進行分光,最後由光電二極體陣列檢測,得到各個組分的吸收信號。經計算機快速處理,得三維立體譜圖。
⑶熒光檢測器
熒光檢測器是一種高靈敏度、高選擇性檢測器。
熒光檢測器的結構及工作原理和熒光光度計相似。
⑷差示折光檢測器
除紫外檢測器之外應用最多的檢測器。
差示折光檢測器是借連續測定流通池中溶液折射率的方法來測定試樣濃度的檢測器。溶液的折射率是純溶劑(流動相)和純溶質(試樣)折射率乘以各物質的濃度之和。因此溶有試樣的流動相和純流動相之間折射率之差表示試樣在流動相中的濃度。
⑸電導檢測器
其作用原理是根據物質在某些介質中電離后所產生電導變化來測定電離物質含量。
高效液相色譜
正相色譜法
採用極性固定相(如聚乙二醇、氨基與腈基鍵合相);流動相為相對非極性的疏水性溶劑(烷烴類如正已烷、環已烷),常加入乙醇、異丙醇、四氫呋喃、三氯甲烷等以調節組分的保留時間。常用於分離中等極性和極性較強的化合物(如酚類、胺類、羰基類及氨基酸類等)。
反相色譜法
一般用非極性固定相(如C18、C8);流動相為水或緩衝液,常加入甲醇、乙腈、異丙醇、丙酮、四氫呋喃等與水互溶的有機溶劑以調節保留時間。適用於分離非極性和極性較弱的化合物。RPC在現代液相色譜中應用最為廣泛,據統計,它占整個HPLC應用的80%左右。
高效液相色譜法,只要求試樣能製成溶液,而不需要氣化,因此不受試樣揮發性的限制。對於高沸點、熱穩定性差、相對分子量大(大於400以上)的有機物(這些物質幾乎佔有機物總數的75%~80%)原則上都可應用高效液相色譜法來進行分離、分析。據統計,在已知化合物中,能用氣相色譜分析的約佔20%,而能用液相色譜分析的約佔70~80%。
1、環境中有機氯農藥殘留量分析
固定相:薄殼型硅膠(37~50mm)
流動相:正己烷
流速:1.5mL/min
色譜柱:50cm´;2.5mm(內徑)
檢測器:差示折光檢測器
可對水果、蔬菜中的農藥殘留量進行分析。
2、稠環芳烴的分析
稠環芳烴多為致癌物質。
固定相:十八烷基硅烷化鍵合相
流動相:20%甲醇-水~100%甲醇;線性梯度淋洗2%/min
流速:1mL/min
柱溫:50℃
柱壓:70´104Pa
檢測器:紫外檢測器
3.陰離子分析
雙柱;薄殼型陰離子交換樹脂分離柱(3×250mm),
流動相:0.003mol·L-1NaHCO3/0.0024mol·L-1Na2CO3,流量138mL/hr。
七種陰離子在20分鐘內基本上得到完全分離,各組分含量在3~50ppm。
發展簡況
液相色譜法開始階段是用大直徑的玻璃管柱在室溫和常壓下用液位差輸送流動相,稱為經典液相色譜法,此方法柱效低、時間長(常有幾個小時)。高效液相色譜法(HighperformanceLiquidChromatography,HPLC)是在經典液相色譜法的基礎上,於60年代後期引入了氣相色譜理論而迅速發展起來的。
它與經典液相色譜法的區別是填料顆粒小而均勻,小顆粒具有高柱效,但會引起高阻力,需用高壓輸送流動相,故又稱高壓液相色譜法(HighPressureLiquidChromatography,HPLC)。又因分析速度快而稱為高速液相色譜法(HighSpeedLiquidChromatography,HSLP)。也稱現代液相色譜。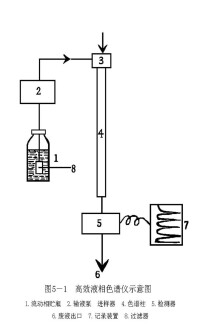
高效液相色譜
