全基因組測序
對未知基因組物種進行個體測序
徠全基因組測序是對未知基因組序列的物種進行個體的基因組測序。 1986年, Renato Dulbecco是最早提出人類基因組測序的科學家之一。他認為如果能夠知道所有人類基因的序列,對癌症的研究將會很有幫助。美國能源部(DOE)與美國國家衛生研究院(NIH),分別在1986年與1987年加入人類基因組計劃。除了美國之外,日本在1981年就已經開始研究相關問題,但是並沒有美國那樣積極。到了1988年,詹姆士·華生(DNA雙螺旋結構發現者之一)成為NIH的基因組部門主管。1990年開始國際合作。1996年,多個國家召開百慕達會議,以2005年完成測序為目標,分配了各國負責的工作,並且宣布研究結果將會及時公布,並完全免費。
①NCI-H209細胞系基因組中,共檢測到22,910個鹼基替換、65個插入缺失(Indels)、58個結構變異;在基因組的編碼區,除了發現RB1 和TP53基因發生點突變和MLL2基因由於發生了G>T的顛換,從而產生了pre-stop codon外,有94個點突變直接改變了氨基酸序列,有36個屬同義突變。
②特定的鹼基及其周圍序列易被煙氣中的多環芳烴和丙烯醛誘變。在NCI-H209細胞系基因組中,G>T/C>A是最為普遍的顛換現象,發生頻率為34%;其次是G>A/C>T(21%)和A>G/T>C(19%);CpG島外的CpG二核苷酸多發生G>T顛換,而CpG島內的CpG二核苷酸多發生G>C顛換,說明煙氣中的致癌物偏好引起甲基化的CpG二核苷酸發生顛換。
③檢測到轉錄偶聯修復(Transcription-coupled repair)和表達相關的修復(Expression-linked repair)在起作用。轉錄偶聯修復作用機制:鳥嘌呤和腺嘌呤上大的加合物是吸煙過程中所釋放的致癌化學物質引起DNA損傷的主要形式,這些大的加合物阻止了轉錄鏈上RNA聚合酶的轉錄過程,而轉錄受阻的RNA聚合酶招募核苷酸剪切修復相關因子對受損的核苷酸進行修復以避免突變發生。在TP53基因突變的肺癌細胞中,G>T顛換常出現在非轉錄鏈,表明在轉錄鏈上相同的損傷已被識別和修復。在本研究中,轉錄鏈上G和A鹼基替換頻率比非轉錄鏈上少,由此看來嘌呤是煙氣致癌物質主要誘變靶標。另外,在NCI-H209細胞系中,轉錄鏈和非轉錄鏈上發生不同類型的突變(G>T、A>G、A>T)兩條鏈基因表達水平也有差異,這就意味著轉錄偶聯修復機制識別、修復不同加合物損傷的能力不同。
表達相關的修復(Expression-linked repair)作用機制:這是一種新的、更為普遍的修復機制,即,高表達的基因中,轉錄鏈及非轉錄鏈的突變頻率都較低。在NCI-H209細胞系中,轉錄鏈和非轉錄鏈上發生G>A的突變,兩條鏈上基因表達水平都很高,這就說明表達相關的修復作用比轉錄偶聯修復作用更為重要。
④在SCLC細胞系中,CHD7基因發生了重排。在NCI-H209細胞系中,CHD7基因3~8外顯子發生連續重複,而另外2個LU-135、NCI-H2171細胞系則攜帶PVT1-CHD7融合基因,說明在肺癌中CHD7基因發生了周期性重排。
以上結果表明,第二代測序技術已成為研究與癌症相關的基因突變過程、細胞損傷修復路徑、基因調控網路的強有力工具。
每個人從受精卵開始就繼承了父母的DNA遺傳信息,並且攜帶一生,不易改變。全基因組測序就是通過運用新一代高通量DNA測序儀,進行10-20倍覆蓋率的個人全基因組測序,然後與人類基因組精確圖譜比較,得到完整的個人全基因組序列,破譯個人全部的遺傳信息的過程。
全基因組測序覆蓋面廣,能檢測個體基因組中的全部遺傳信息;準確性高,其準確率可高達99.99%。
全基因組測序揭示了人類生、老、病、死的奧秘,使人類從根本上認知疾病發生的原因,做到正確的治療疾病、儘早的預防疾病。
提取基因組DNA,然後隨機打斷,電泳回收所需長度的DNA片段(0.2~5kb),加上接頭,進行基因簇cluster製備或電子擴增E-PCR,最後利用Paired-End(Solexa)或者Mate-Pair(SOLiD)的方法對插入片段進行測序。然後對測得的序列組裝成Contig,通過Paired-End的距離可進一步組裝成Scaffold,進而可組裝成染色體等。組裝效果與測序深度與覆蓋度、測序質量等有關。目前常用的組裝有:SOAPdenovo、Trimity、Abyss等。
雙末端(Paired-End)測序原理
測序深度(Sequencing Depth):測序得到的鹼基總量(bp)與基因組大小(Genome)的比值,它是評價測序量的指標之一。測序深度與基因組覆蓋度之間是一個正相關的關係,測序帶來的錯誤率或假陽性結果會隨著測序深度的提升而下降。重測序的個體,如果採用的是雙末端或Mate-Pair方案,當測序深度在10~15X以上時,基因組覆蓋度和測序錯誤率控制均得以保證。
測序深度對基因組覆蓋度和測序錯誤率的影響
(HOM:純合體 HET:雜合體)
全基因組重測序的個體,通過序列比對,可以找到大量的單核苷酸多態性(SNP),插入缺失(InDel,Insertion/Deletion)和結構變異(SV,Structure Variation)位點。SBC可以協助客戶,通過生物信息手段,分析不同個體基因組間的結構差異,同時完成SNP及基因組結構註釋。
測序覆蓋度:基因組被測序得到的鹼基覆蓋的比例;測序覆蓋度是反映測序隨機性的指標之一;測序序深度與覆蓋度之間的關係可以過Lander-Waterman Model(1988)來確定。當深度達到5X時,則可覆蓋基因組的約99.4%以上。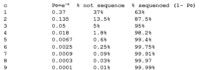
徠全基因組測序
1.數據量產出
總鹼基數量、Total Mapping Reads、Uniquely Mapping Reads統計,測序深度分析。
2.一致性序列組裝
與參考基因組序列(Reference genome sequence)的比對分析,利用貝葉斯統計模型檢測出每個鹼基位點的最大可能性基因型,並組裝出該個體基因組的一致序列。
3.SNP檢測及在基因組中的分佈
提取全基因組中所有多態性位點,結合質量值、測序深度、重複性等因素作進一步的過濾篩選,最終得到可信度高的SNP數據集。並根據參考基因組信息對檢測到的變異進行註釋。
4.InDel檢測及在基因組的分佈
在進行mapping的過程中,進行容gap的比對並檢測可信的short InDel。在檢測過程中,gap的長度為1~5個鹼基。對於每個InDel的檢測,至少需要3個Paired-End序列的支持。
5.Structure Variation檢測及在基因組中的分佈
SBC能夠檢測到的結構變異類型主要有:插入、缺失、複製、倒位、易位等。根據測序個體序列與參考基因組序列比對分析結果,檢測全基因組水平的結構變異並對檢測到的變異進行註釋。
基本信息
- 中文名
- 全基因組測序
- 測序目標
- 2005年
- 準備時間
- 19世紀80-90年代
- 國際合作
- 1990年