溶酶體酶
溶酶體酶
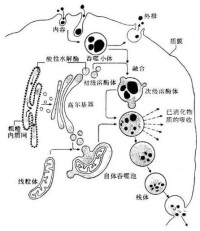
溶酶體(lysosome)為細胞漿內由單層脂蛋白膜包繞的內含一系列酸性水解酶的小體。是細胞內具有單層膜囊狀結構的細胞器,溶酶體內含有許多種水解酶類,能夠分解很多種物質,溶酶體被比喻為細胞內的“酶倉庫”“消化系統”。
溶酶體酶
現已知各類細胞的溶酶體中約含60種酶,包括蛋白質、糖類、脂類等物質的水解酶類,如酸性磷酸脂酶、組織蛋白酶、核糖核酸酶以及芳香基硫酸脂酶A和B等。各類溶酶體所含水解酶也有所不同,大多數溶酶體里的酶是糖蛋白,但也有例外,如鼠肝細胞和腎細胞溶酶體里的酶大部分是脂蛋白。常見的溶酶體酶包括:芳基硫酸酯酶A(ASA)、半乳糖腦苷脂酶、β-半乳糖苷酶、β-氨基己糖苷酶A、總氨基己糖苷酶、β-葡萄糖苷酶、神經鞘磷脂酶、α-半乳糖苷酶、α-葡糖苷酶、α-岩藻糖苷酶、α-甘露糖苷酶、酸性型β-甘露糖苷酶、天冬氨醯氨基葡糖苷酶、α-L-艾杜糖苷酸酶、艾杜糖醛酸硫酸酯酶、乙醯肝素-N-硫酸酯酶、α-N-乙醯氨基葡糖苷酶、乙醯CoA-氨基葡糖-N-乙醯轉移酶、N-乙醯氨基葡糖-6-硫酸酯酶、半乳糖-6-硫酸酯酶、β-半乳糖苷酶、芳基硫酸酯酶、β-葡糖醛酸苷酶、透明質酸酶、磷酸轉移酶、棕櫚醯蛋白硫脂酶、羧肽酶(TPP1溶酶體肽酶)、神經醯胺酶、唾液酸酶、酸性酯酶等等。
溶酶體內的酶活性不足(主要是酸性水解酶)、激活蛋白、轉運蛋白或溶酶體蛋白加工校正酶的缺乏而引起溶酶體功能缺陷,造成次級溶酶體內相應底物不能被消化,底物積蓄,代謝障礙,形成貯積性疾病,稱為溶酶體貯積症,溶酶體貯積症不僅影響機體某個器官的正常功能,往往也會影響到整個機體代謝活動的協調性,引起多種疾病。目前已知此類疾病有40種以上,大致可分為糖原累積病、腦苷脂沉積病、台-薩氏綜合征和粘多糖沉積病等幾大類。
糖原累積病
糖原累積病(glycogen storage disease,GSD),發病原因是肝和肌細胞的溶酶體缺乏一種酸性Ⅱ一葡萄糖苷酶。正常時此酶分解糖原,而此酶缺乏時,溶酶體吞噬的過剩糖原無法降解,大量堆積在次級溶酶體內使其腫脹,最終導致溶酶體破裂,其他酶漏出,嚴重破壞組織細胞。此病屬常染色體缺陷性遺傳病,患者多為小孩,常在兩周歲以前死亡。
腦苷脂沉積病
腦苷脂沉積病(cerebrosidosis),又名Gaucher病(戈謝病),是巨噬細胞和腦神經細胞的溶酶體缺乏β-葡萄糖苷酶(β-glucocerehrosidase)造成的。大量的葡萄糖腦苷脂沉積在這些細胞溶酶體內,巨噬細胞變成Gaucher細胞,患者發生肝、脾腫大,血小板減少,貧血,骨痛等癥狀,嚴重的發生眼球運動障礙,共濟失調等中樞神經系統癥狀。此病多發生於各個年齡段,從嬰幼兒到成人均可發病,癥狀輕重差異大,發病越早,癥狀越重,則預后越差。。
台-薩氏綜合征
台-薩氏綜合征(Tay-Sachs diesease),溶酶體缺少氨基已糖酯酶A(β-N-hexosaminidase),後果是GM2神經節苷脂不能水解而貯積在溶酶體中,從而使細胞發生功能損害。該病在德系猶太人中發病率最高,我國極少見。表現為家族性痴獃,大腦黃斑變性。本病以神經細胞受損較明顯,因此,神經組織功能障礙很突出,表現為漸進性失明、痴獃和癱瘓。我國有報道的病例至今只有兩例。
粘多糖沉積病
粘多糖沉積病(mucopolysaccharidosis)是一組黏多糖進行性代謝障礙的遺傳病。病理是溶酶體內缺乏黏多糖降解酶,因而不能分解黏多糖類,使這些物質堆積在次級溶酶體內。患者面容粗獷,骨骼異常,智力發育不全,內臟功能普遍受損,角膜混濁。患兒體內有糖胺聚糖沉積,以腦、心、肝、脾較為顯著,角膜、骨、肌腱次之,心臟受累以瓣膜、心肌為主。臨床表現也有差異,目前共分為7型。
溶酶體酶缺失、不足或活性差可以採用溶酶體酶活性檢測的方法進行測量,溶酶體酶活性檢測是確診酶體貯積症的金標準。國內有北京協和醫院、上海新華醫院、北京中科醫學檢驗所等單位可以進行溶酶體酶活性的檢測。