注射用蘭索拉唑
注射用蘭索拉唑
注射用蘭索拉唑
化學結構式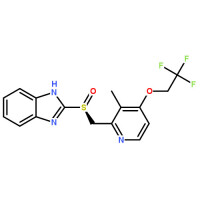 本品主要成份為蘭索拉唑。
本品主要成份為蘭索拉唑。
化學結構式
化學名稱:2-[[[3-甲基-4-(2,2,2-三氟乙氧基)-2-吡啶基]甲基]亞磺醯基-1H-苯並咪唑。
化學結構式:
分子式:C16H14F3N3O2S
分子量:369.36
本品為白色或類白色疏鬆塊狀物或粉末。
用於口服療法不適用的伴有出血的十二指腸潰瘍。
30mg。
靜脈滴註:通常成年人一次30mg,用0.9%氯化鈉注射液100ml溶解后,一日2次,推薦靜滴時間30分鐘,療程不超過7天。使用時注意:
(1)本品靜滴使用時應配有孔徑為1.2μm的過濾器,以便去除輸液過程中可能產生的沉澱物。這些沉澱物有可能引起小血管栓塞而產生嚴重後果。
(2)在噴出性或湧出性大量出血、血管暴露等危險性大的情況下,應先採用內窺鏡下止血措施。
(3)本品僅用於靜脈滴注。溶解后應儘快使用,勿保存。避免與0.9%氯化鈉注射液以外的液體和其他藥物混合靜滴。
(4)經本品治療的前3日內達到止血效果的,應改用口服用藥,不可無限制靜脈給葯。
國內臨床研究中,本品一日2次,每次30mg靜脈滴注,5天療程。127例受試者中發生4例(3.64%)不良反應,主要是白細胞減少(1.82%)、轉氨酶輕度升高(0.91%)和皮疹(0.91%)。白細胞減少者一周后複查正常,轉氨酶輕度升高者十天後複查正常。
國外上市后超過1000名患者使用注射用蘭索拉唑後有較好的耐受性。在美國4個臨床試驗中有161名患者使用注射用蘭索拉唑,超過1%的不良反應有噁心(1.3%)、頭痛(1%)、注射部位痛感(1%);低於1%的不良反應有腹痛、腹瀉、消化不良、嘔吐、頭暈、感覺異常、味覺異常、皮疹和血管擴張。未見與口服給葯不同的不良反應。
日本221例受試者使用注射用蘭索拉唑的臨床研究資料報道,發生31例(14.0%)臨床實驗室檢查值異常,主要為ALT升高(6.2%)、AST升高(5.7%)、LDH升高(2.0%)、γ-GTP升高(1.5%)等檢查值異常變化。
以下不良反應為口服蘭索拉唑所見,但靜脈注射也有可能發生:
1.治療時應密切觀察,如有下列嚴重的不良反應,應及時中止用藥和處理。
(1)出現過敏反應(全身出疹、面部浮腫、呼吸困難等)(<0.1%),甚至引起休克(<0.1%);
(4)中毒性表皮壞死溶解症(Lyell綜合征)、皮膚粘膜眼綜合征(stevens-Johnson綜合征)(<0.1%);
2.其他的不良反應;出現以下不良反應時中止用藥,必要時進行適當的處理。
1.對蘭索拉唑及處方中任一成分過敏的患者禁止使用本品。
2.正在使用硫酸阿扎那韋的患者禁止使用本品。
1、以下患者慎重用藥:
(1)有藥物過敏症既往史的患者;
(2)肝損傷的患者(因本葯的代謝、排泄延遲);
2、本品治療會掩蓋消化道腫瘤的癥狀,應排除惡性腫瘤後方可用藥。
3、本品治療時密切觀察病情,治療無效時應改用其它療法。
4、本品目前尚無超過7日的用藥經驗。
5、同類質子泵抑製藥物奧美拉唑(omeprazole)在國外有導致視力損害的報道,本品尚不清楚。
6、動物實驗中,大鼠長期大量使用本品后,出現良性睾丸間質細胞腫瘤,類癌瘤與視網膜萎縮。但類似現象在小鼠的致癌性試驗、犬、猴的毒性試驗中未出現。
大鼠口服蘭索拉唑的試驗中可見胎仔血漿中蘭索拉唑藥物濃度比母體血漿中藥物濃度高;另外,兔(口服30mg/kg/日)試驗中可見胎仔死亡率的增加。對孕婦和可能妊娠的婦女,建議只有在判斷治療的益處大於風險時方可使用本品。大鼠口服蘭索拉唑的試驗中,蘭索拉唑可經母乳分泌。建議哺乳期婦女盡量避免使用本品,必須用藥時,應停止哺乳。
兒童使用本品的安全性尚未確定,尚無使用經驗。
一般老年人生理機能下降,故應慎重用藥。
蘭索拉唑通過肝臟細胞色素P450酶系統代謝,特別是CYP3A4和CYP2C19酶代謝。蘭索拉唑可顯著地、長時間抑制胃酸分泌,故理論上可促進或抑制一些合用藥物的吸收。
目前無藥物過量的臨床經驗供參考,但蘭索拉唑不能通過血液透析從循環系統中清除。
藥理作用:
蘭索拉唑屬於質子泵抑製劑。本葯分佈於胃粘膜壁細胞的酸性環境后,轉變為有活性的代謝物。這種代謝物與存在於酸生成部位的H+,K+-ATP酶的巰基結合,通過抑制H+,K+-ATP酶的活性而抑制酸分泌。
蘭索拉唑抑制胃酸分泌作用呈劑量依賴性,葯后24小時內對基礎和刺激引起的胃酸分泌均有抑制作用。健康成人1次30mg,一日2次靜脈給葯,可見持續的胃酸分泌抑制作用。
有報道在酸性條件下,血液凝固與血小板聚集受到很大損害。血液凝固所形成的纖維蛋白,在酸性條件下可被胃蛋白酶溶解。本葯通過升高胃內pH值而改善血液凝固與血小板聚集功能,抑制胃蛋白酶的活性而發揮抑制出血的作用。另外,在酸性條件下,胃的損傷粘膜的修復受到抑制,本葯通過抑制酸分泌而使胃內pH值上升,促進損傷粘膜的修復。
毒理研究:
遺傳毒性:Ames試驗。大鼠肝細胞程序外DNA合成試驗以及小鼠微核試驗、大鼠骨髓細胞染色體畸變試驗結果均為陰性。體外人淋巴細胞染色體畸變實驗結果為陽性。
生殖毒性:大鼠和家兔靜脈給予蘭索拉唑劑量達30mg/kg/天(按體表面積計算相當於人臨床推薦劑量的8倍和16倍),未發現對生育力和胚胎有不良影響。
致癌研究:SD大鼠連續24個月口服蘭索拉唑5-150mg/kg/天(按體表面積計算相當於體重為5公斤患者的臨床推薦劑量30mg/d的1~40倍),結果顯示蘭索拉唑以劑量依賴方式誘發胃腸嗜鉻樣(ECL)細胞增生和良性ECL細胞瘤;受試動物胃上皮出現腸上皮化的發生率增加。雄性大鼠睾丸間質細胞腺瘤發生率以劑量相關方式增加,給藥劑量為15~150mg/kg/天(按體表面積計算相當於人臨床推薦劑量的4~40倍)的大鼠,這些腺瘤的發生率超過該種系大鼠的自然發生率(1.4~10%)。一年毒性研究中,給藥劑量為50mg/kg/天(按體表面積計算相當於人臨床推薦劑量13倍)的30隻大鼠中,有1隻出現睾丸間質細胞腺瘤。CD-1小鼠連續24個月口服蘭索拉唑15~600mg/kg/天(按體表面積計算相當於人臨床推薦劑量的2~80倍),結果蘭索拉唑以劑量依賴方式誘發ECL細胞增生,給葯小鼠肝臟腫瘤發生率升高,300和600mg/kg/天組雄性小鼠和150~600mg/kg/天組雌性小鼠腫瘤的發生率超過了該種系小鼠歷史腫瘤的發生率範圍。給予蘭索拉唑75~600mg/kg/天,小鼠發生睾丸腺瘤。
吸收蘭索拉唑靜脈給葯時,血清中濃度存在個體差異。健康人靜脈注射蘭索拉唑30mg(30min),血葯濃度呈雙指數降低,終末半衰期為1.3(±0.5)h,血葯峰濃度(Cmax)為1705(±292)ng/ml,AUC為3192(±1745)ng•h/ml,每日一次口服或靜脈注射蘭索拉唑30mg,7天後的葯代學參數不隨時間變化而變化。
口服蘭索拉唑研究提示性別和腎功能不全患者的葯代無差異,因此靜脈用藥無需調整給藥劑量。
代謝蘭索拉唑的血漿消除半衰期與抑制胃酸分泌的作用時間無關。蘭索拉唑的消除半衰期低於2小時,而抑制胃酸分泌的作用時間至少在24小時以上。蘭索拉唑通過細胞色素P450酶系統代謝,特別是CYP2C19酶和CYP3A4酶代謝。據報道,CYP2C19存在遺傳多態性,亞洲系蒙古人種約有10~20%為慢代謝者型。蘭索拉唑在肝內廣泛代謝,血漿中可檢測到兩個主要代謝產物(羥基化亞磺醯基和磺基衍生物)。這些代謝物只在胃壁細胞小管內轉變為2個通過抑制H+,K+-ATP酶的活性成分,但它們在體循環中測不出。
消除靜脈注射的蘭索拉唑平均清除率為11.1(±3.8)L/h。健康成年男子,1次靜脈給葯30mg,尿中未見原形葯,全部為代謝產物,至給葯結束24小時后的尿中累積排泄率為12~17%。一項口服14C標記的蘭索拉唑研究顯示,約1/3的給藥量(放射活性)通過尿排泄,約2/3的放射性物質在糞便中出現,提示蘭索拉唑的代謝產物通過膽汁和尿排泄。
遮光,密閉保存。
(1)西林瓶1瓶/盒。
(2)西林瓶2瓶/盒。
24個月。
國家食品藥品監督管理局標準YBH02092012
