范可尼貧血
范可尼貧血
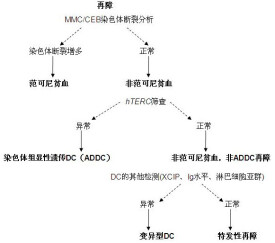
范可尼貧血(Fanconi anemia)是一種罕見的常染色體隱性遺傳性血液系統疾病,屬於先天性再障,稱為Fanconi貧血(范可尼貧血),這類病人除有典型再障表現外,還伴有多發性的先天畸形(皮膚棕色色素沉著,骨骼畸形、性發育不全等)。其病因可能是染色體的異常。同時,還會伴有精子減少等其它特徵。經常與其它疾病伴發,因此科學家一直在探索這個疾病的根本病因。
科學家們發現,在范可尼貧血病人中,因缺少DNA螺旋的一個關鍵基因BRIP1,因此許多與它相互作用的基因便不能發揮功能,從而引發一系列伴隨疾病或癥狀,如BRCA1這個基因無法作用於DNA轉錄,因此導致它經常與腫瘤伴發。最新的揭示了范可尼貧血發病機理,以及DNA修復的關係,為治療該疾病提供了充分的遺傳學依據。
2018年5月11日,國家衛生健康委員會等5部門聯合制定了《第一批罕見病目錄》,范可尼貧血被收錄其中。
1. 貧血的一般表現,出血傾向及易感染。多見皮膚色素沉著,或片狀棕色斑,體格、智力可發育落後。無肝、脾、淋巴結腫大。
(一)發病年齡 多於兒童期發病。男孩以4~7歲、女孩以6~10歲發病者較多。
(二)家族史 約10%~30%父母為近親結婚。
(三)實驗室檢查
1. 血象 典型表現為全血細胞減少。多先有血小板減少,逐漸發展為全血細胞減少。少數病例可僅一系或兩系細胞減少。貧血可呈大細胞性。網織紅細胞減少。粒細胞可見中毒顆粒。
3. 生化改變 約半數患兒出現氨基酸尿(多為脯氨酸尿),胎兒血紅蛋白增多(5%~15%)。紅細胞長期存在( 抗原(正常2歲前消失)。
4. 染色體異常 外周血淋巴細胞培養作染色體分析可見斷裂、單體交換、環形染色體等畸變。姐妹染色體交換減少更具診斷意義。
(一)一般支持療法 同再生障礙性貧血。貧血重時應予輸血。
(二)激素療法 給予一種雄激素及一種皮質激素多數病例有效。常用羥甲烯龍[最多可用至5mg/(kg?d)]加較小量潑尼松[lmg/(kg?d)],一般於用藥后2~4個月即有明顯生血反應,用至血紅蛋白達正常水平后逐漸減量,以維持量維持血紅蛋白在正常低限水平。治療期間應注意肝功損害等毒副作用問題。
(三)有條件者可做骨髓移植。
未用雄激素治療前,一旦出現貧血,5年生存率約為50%。經用激素治療後生存率大為提高,但有5%~10%病例發生急性粒細胞白血病或其他惡性腫瘤。
2020年03月06日,荷蘭皇家科學院Hubrecht研究所和英國MRC分子生物學實驗室的科學家,在頂級期刊《自然》上發表的研究成果,他們發現了一個全新的DNA修復機制,可以安全地修復酒精代謝物乙醛導致的DNA損傷。如果能找到激活這條修復通路的方法,或許可以用於治療范可尼貧血(FA)。
基本信息
- 中文名
- 范可尼貧血
- 癥狀表現
- 多種多樣的肢體和(或)器官畸形和進行性骨髓衰竭
- 就診科室
- 內科