轉染
轉染
轉染(transfection)是真核細胞主動或被動導入外源DNA片段而獲得新的表型的過程。從本質上講,和轉化沒有根本的區別。無論是轉染還是轉化,其關鍵因素都是用氯化鈣處理大腸桿菌細胞,以提高細胞膜的通透性,從而使外源DNA分子能夠容易進入細胞內部。所以在習慣上,人們往往也通稱轉染為廣義的轉化。常規轉染技術可分為瞬時轉染和穩定轉染(永久轉染)兩大類。
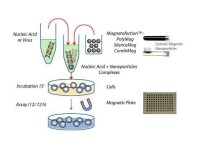
細胞化學轉染
常規轉染技術可分為兩大類,一類是瞬時轉染,一類是穩定轉染(永久轉染)。前者外源DNA/RNA不整合到宿主染色體中,因此一個宿主細胞中可存在多個拷貝數,產生高水平的表達,但通常只持續幾天,多用於啟動子和其它調控元件的分析。一般來說,超螺旋質粒DNA轉染效率較高,在轉染后24-72小時內(依賴於各種不同的構建)分析結果,常常用到一些報告系統如熒光蛋白,β半乳糖苷酶等來幫助檢測。後者也稱穩定轉染,外源DNA既可以整合到宿主染色體中,也可能作為一種遊離體(episome)存在。儘管線性DNA比超螺旋DNA轉入量低但整合率高。外源DNA整合到染色體中概率很小,大約1/104轉染細胞能整合,通常需要通過一些選擇性標記,如來氨丙基轉移酶(APH;新黴素抗性基因),潮黴素B磷酸轉移酶(HPH),胸苷激酶(TK)等反覆篩選,得到穩定轉染的同源細胞系。
轉染技術的選擇對轉染結果影響也很大,許多轉染方法需要優化DNA與轉染試劑比例,細胞數量,培養及檢測時間等。
包括:
1.DEAE-葡聚糖法
轉染示意圖
2.磷酸鈣法
磷酸鈣法是磷酸鈣共沉澱轉染法,因為試劑易取得,價格便宜而被廣泛用於瞬時轉染和穩定轉染的研究,先將DNA和氯化鈣混合,然後加入到PBS中慢慢形成DNA磷酸鈣沉澱,最後把含有沉澱的混懸液加到培養的細胞上,通過細胞胞膜的內吞作用攝入DNA。磷酸鈣似乎還通過抑制血清中和細胞內的核酸酶活性而保護外源DNA免受降解.
3.人工脂質體法。
人工脂質體法採用陽離子脂質體,具有較高的轉染效率,不但可以轉染其他化學方法不易轉染的細胞系,而且還能轉染從寡核苷酸到人工酵母染色體不同長度的DNA,以及RNA,和蛋白質。此外,脂質體體外轉染同時適用於瞬時表達和穩定表達,與以往不同的是脂質體還可以介導DNA和RNA轉入動物和人的體內用於基因治療。LipoFiter 脂質體轉染試劑(LipoFiterLiposomalTransfectionReagent)是一種適合於把質粒或其它形式的核酸,以及核酸蛋白複合物轉染到培養的真核細胞中的高效陽離子脂質體轉染試劑。它可以和帶負電荷的核酸結合后形成複合物,當複合物接近細胞膜時被內吞成為內體進入細胞質,隨後DNA複合物被釋放進入細胞核內,至於DNA是如何穿過核膜的,其機理目前還不十分清楚。
包括:①顯微注射②電穿孔③基因槍等,顯微注射雖然費力,但是非常有效的將核酸導入細胞或細胞核的方法。這種方法常用來製備轉基因動物,但卻不適用於需要大量轉染細胞的研究。電穿孔法常用來轉染如植物原生質體這樣的常規方法不容易轉染的細胞。電穿孔靠脈衝電流在細胞膜上打孔而將核酸導入細胞內。導入的效率與脈衝的強度和持續時間有關係,基因槍依靠攜帶了核酸的高速粒子而將核酸導入細胞內,這種方法適用於培養的細胞核在體的細胞。
細胞轉染實驗
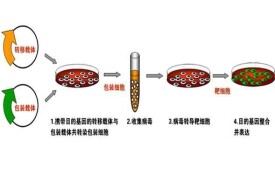
基因轉染示意圖
外源基因進入細胞主要有四種方法:電擊法、磷酸鈣法、脂質體介導法和病毒介導法。電擊法是在細胞上短時間暫時性的穿孔讓外源質粒進入;磷酸鈣法和脂質體法是利用不同的載體物質攜帶質粒通過直接穿膜或者膜融合的方法使得外源基因進入細胞;病毒法是利用包裝了外源基因的病毒感染細胞的方法使得其進入細胞。但是由於電擊法和磷酸鈣法的實驗條件控制較嚴、難度較大;病毒法的前期準備較複雜、而且可能對於細胞有較大影響;所以現在對於很多普通細胞系,一般的瞬時轉染方法多採用脂質體法。
利用脂質體轉染法最重要的就是防止其毒性,因此脂質體與質粒的比例,細胞密度以及轉染的時間長短和培養基中血清的含量都是影響轉染效率的重要問題,通過實驗摸索的合適轉染條件對於效率的提高有巨大的作用。
(1)材料
293T細胞、MyoD表達質粒和EGFP表達質粒、DMEM培養基、鏈黴素/青霉素(雙抗)、FCS(小牛血清)、PBS(磷酸鹽緩衝溶液)、胰酶/EDTA消化液、轉染試劑(TransFast)
(2)器材
20ul/200ul/1ml微量移液器和Tip頭酒精燈、廢液缸、血球計數板、渦旋振蕩器、恆溫水浴箱、台式離心機、35mm培養皿、轉染管、15ml離心管、觀察用倒置顯微鏡熒光顯微鏡和CCD
細胞傳代
(1)試驗準備:200ul/1mlTip頭各一盒(以上物品均需高壓滅菌),酒精棉球,廢液缸,試管架,微量移液器,記號筆,培養皿,離心管。
(2)棄掉培養皿中的培養基,用1ml的PBS溶液洗滌兩次。
(3)用Tip頭加入1mlTrypsin液,消化1分鐘(37 C,5%CO)。用手輕拍培養瓶壁,觀察到細胞完全從壁上脫落下來為止。
(4)加入1ml的含血清培養基終止反應。
(5)用Tip頭多次吹吸,使細胞完全分散開。
(6)將培養液裝入離心管中,1000rpm離心5min。
(7)用培養液重懸細胞,細胞計數后選擇0.8X10 個細胞加入一個35mm培養皿。
(8)將合適體積完全培養液加入離心管中,混勻細胞后輕輕加入培養皿中,使其均勻分佈。
(9)將培養皿轉入CO培養箱中培養,第二天轉染。
細胞轉染
1)轉染試劑的準備
A.將400ul去核酸酶水加入管中,震蕩10秒鐘,溶解脂狀物。
B.震蕩后將試劑放在-20攝氏度保存,使用前還需震蕩。
2)選擇合適的混合比例(1:1-1:2/脂質體體積:DNA質量)來轉染細胞。在一個轉染管中加入合適體積的無血清培養基。加入合適質量的MyoD或者EGFP的DNA,震蕩后在加入合適體積的轉染試劑,再次震蕩。
5)將混合液在室溫放置10—15分鐘。
6)吸去培養板中的培養基,用PBS或者無血清培養基清洗一次。
7)加入混合液,將細胞放回培養箱中培養一個小時。
8)到時后,根據細胞種類決定是否移除混合液,之後加入完全培養基繼續培養24-48小時。
第二次細胞傳代
1)在轉染后24小時,觀察實驗結果並記錄綠色熒光蛋白表達情況。
2)再次進行細胞傳代,按照免疫染色合適的密度0.8X105個細胞/35mm培養皿將細胞重新放入培養皿中。
3)在正常條件下培養24小時后按照染色要求條件固定。
轉染細胞篩選
1.確定抗生素作用的最佳濃度:
不同的細胞株對各種抗生素有不同的敏感性,因此在篩選前要做預試驗,確定抗生素對所選擇細胞的最低作用濃度。
1) 提前24 小時在96 孔板或24 孔板中接種細胞8 孔,接種量以第二天長成25%單層為宜,置CO2 孵箱中37℃培養過夜。
2) 將培養液換成含抗生素的培養基,抗生素濃度按梯度遞增(0, 50, 100, 200,400, 600, 800 和1000μg/ml)。
3) 培養10-14 天以絕大部分細胞死亡濃度為準,一般為400-800μg/ml,篩選穩定表達克隆時可比該濃度適當提高一個級別維持時使用篩選濃度的一半.
2.轉染按前面的步驟進行。
3.轉染72 小時后按1:10 的比例將轉染細胞在6 孔板中傳代,換為含預試驗中確定的抗生素濃度的選擇培養基。在6 孔板內可見單個細胞,繼續培養可見單個細胞分裂繁殖形成單個抗性集落,此時可用兩種方法挑選單克隆。
1) 濾紙片法:用消毒的5x5mm 濾紙片浸過胰酶,將濾紙片貼在單細胞集落上10-15 秒,取出粘附有細胞的濾紙片放於24 孔板中繼續加壓培養。細胞在24 孔板中長滿後轉入25cm 培養瓶中,長滿后再轉入75cm 培養瓶中培養。
2) 有限稀釋法:將細胞消化下來后做連續的10 倍稀釋(10-2—10-10),將每一稀釋度的細胞滴加到96 孔板中培養,7-10 天后,選擇單個克隆生長的孔再一次進行克隆。
4. ELISA 或Western blot 檢測單克隆細胞中外源蛋白的表達情況由於不同克隆的表達水平存在差異因此可同時挑選多個克隆選擇表達量最高的克隆傳代並保種。

轉染試劑
線型PEI(LinePEI,LPEI)與其衍生物用作基因轉染載體的研究比分枝狀PEI(BranchedPEI,BPEI)要早一些,過去的研究認為在不考慮具體條件,LPEI/DNA轉染複合物的細胞毒性較低,有利於細胞定位,因此與BPEI相比應該轉染效率高一些。但最近研究表明BPEI的分枝度高有利於形成小的轉染複合物,從而提高轉染效率,但同時細胞毒性也增大。超高分枝的、較柔性的PEI衍生物含有額外的仲胺基和叔胺基,在染實驗中發現這種PEI的毒性低,但轉染效率卻較高。
有的產品採用各種分枝狀和超高分枝狀的小分子PEI與各種含有生理條件下可降解鍵的交聯劑交聯,合成出的一系列高分枝的可降解的PEI衍生物。聚合物的分枝結構使得其具有較高的正電性,因此易於高效地包裹各種DNA、RNA分子及質粒形成小的納米顆粒,從而提高轉染效率,當所形成複合物進入細胞以後,其中所含的生理條件下可降解的化學鍵在細胞內水解,使交聯聚合物分解為無細胞毒性的小分子PEI,這樣結構的轉染試劑在體外應用可以獲得高的轉染效率和低的細胞毒性,其可降解性對體內應用也具有重要的意義。
現在最新的轉染試劑是採用納米材料製作,如Engreen,其原理是:分子內含有許多氨基,在生理PH下會發生質子化,這些質子化的氨基可以中和DNA質粒表面的負電荷,使DNA分子由伸展結構壓縮為體積相對較小的DNA粒子,並包裹在其中,使DNA免受核酸酶的降解。轉染複合物主要是通過細胞內吞作用將DNA轉移進入細胞,形成內含體(endosome),DNA從內含體釋放,進入細胞質中,再進一步進入核內轉錄、表達。通過納米技術生產出的轉染試劑在納米尺度表現出結合保護DNA能力強、毒性低的獨特性能。
基本信息
- 中文名
- 轉染
- 外文名
- transfection
- 分類
- 瞬時轉染和穩定轉染
- 學科
- 生物學