遺傳性大皰性表皮鬆解症
遺傳性大皰性表皮鬆解症
大皰性表皮鬆解症(epidermolysis bullosa,EB)分為遺傳性和獲得性兩種。遺傳性EB至少包括23種不同類型,依據發病部位不同可分為三類: ①單純性大皰性表皮鬆解症(simplexEB,EBS),水皰在表皮內 ②交界性大皰性表皮鬆解症(junctionalEB,JEB),水皰發生於透明層; ③營養不良性大皰性表皮鬆解症(dystrophic EB,DEB),水皰發生在緻密下層。 2018年5月11日,國家衛生健康委員會等5部門聯合制定了《第一批罕見病目錄》,遺傳性大皰性表皮鬆解症被收錄其中。
【病因和發病機制】
已經證實EBS與編碼角蛋白5和14的基因突變有關;JEB與編碼板層素5、xⅦ型膠原等物質的基因突變有關;DEB與編碼Ⅶ型膠原的基因突變有關。由於編碼表皮和基底膜帶結構蛋白成分的基因突變,使這些蛋白合成障礙或結構異常,導致不同解剖部位水皰的產生。
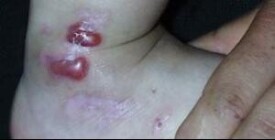
各型大皰性表皮鬆解症的共同特點是皮膚在受到輕微摩擦或碰撞后就出現水皰及血皰(圖25—4)。好發於肢端及四肢關節伸側,嚴重者可累及任何部位。皮榻龠合后可形成瘢痕或粟丘疹,肢端反覆發作的皮損可使指趾甲脫落。
單純型僅累及肢端及四肢關節伸側,不累及黏膜,皮損最表淺,愈后一般不留瘢痕。營養不良型可累及任何部位(包括黏膜),病情多較重,常在出生后即出現皮損,且位置較深,癒合后遺留明顯的瘢痕,肢端反覆發生的水皰及瘢痕可使指趾間的皮膚粘連,指骨萎縮形成爪形手;口咽部黏膜反覆潰破、結痂可致張口、吞咽困難,愈后不佳. 交界型罕見,出生后即出現廣泛水皰、大皰及糜爛面,預后差,多在2歲內死亡。
【診斷和鑒別診斷】根據家族史、臨床特點,結合免疫組化及透射電鏡檢查一般可以確診及分型。
【治療】目前尚無特效療法。應保護皮膚,防止摩擦和壓迫,可用非粘連性合成敷料、無菌紗布或廣譜抗生素軟膏防治感染,同時應加強支持療法。