分子動力學
結合物理、數學和化學的綜合技術
分子動力學是一門結合物理,數學和化學的綜合技術。分子動力學是一套分子模擬方法,該方法主要是依靠牛頓力學來模擬分子體系的運動,以在由分子體系的不同狀態構成的系綜中抽取樣本,從而計算體系的構型積分,並以構型積分的結果為基礎進一步計算體系的熱力學量和其他宏觀性質。
1957年:基於剛球勢的分子動力學法(AlderandWainwright)
分子動力學
1971年:剛體系への拡張(RahmanandStillinger)
1977年:約束動力學方法(Rychaert等)
1980年:恆壓條件下的動力學方法(Andersenの方法、Parrinello-Rahman法)
1983年:非平衡態動力學方法(GillanandDixon)
1984年:恆溫條件下的動力學方法(能勢‐フーバーの方法)
1985年:第一原理分子動力學法(→カー・パリネロ法)
1991年:巨正則系綜的分子動力學方法(CaginandPettit)
確定起始構型
進行分子動力學模擬的第一步是確定起始構型,一個能量較低的起始構型是進行分子模擬的基礎,一般分子的起始構型主要來自實驗數據或量子化學計算。
在確定起始構型之後要賦予構成分子的各個原子速度,這一速度是根據波爾茲曼分佈隨機生成的,由於速度的分佈符合波爾茲曼統計,因此在這個階段,體系的溫度是恆定的。另外,在隨機生成各個原子的運動速度之後須進行調整,使得體系總體在各個方向上的動量之和為零,即保證體系沒有平動位移。
進入平衡相
由上一步確定的分子組建平衡相,在構建平衡相的時候會對構型、溫度等參數加以監控。
進入生產相
進入生產相之後體系中的分子和分子中的原子開始根據初始速度運動,可以想象其間會發生吸引、排斥乃至碰
分子動力學
步驟
以下是做模擬的一般性步驟,具體的步驟和過程依賴於確定的系統或者是軟體,但這不影響我們把它當成一個入門指南:
1)首先我們需要對我們所要模擬的系統做一個簡單的評估, 三個問題是我們必須要明確的:
做什麼(what to do)為什麼做(why to do)怎麼做(how to do)
2)選擇合適的模擬工具,大前提是它能夠實現你所感興趣的目標,這需要你非常謹慎的查閱文獻,看看別人用這個工具都做了些什麼,有沒有和你相關的,千萬不要做到一半才發現原來這個工具根本就不能實現你所感興趣的idea,切記!
考慮1:軟體的選擇,這通常和軟體主流使用的力場有關,而軟體本身就具體一定的偏向性,比如說,做蛋白體系,Gromacs,Amber,Namd均可;做DNA, RNA體系,首選肯定是Amber;做界面體系,Dl_POLY比較強大,另外做材料體系,Lammps會是一個不錯的選擇
考慮2:力場的選擇。力場是來描述體系中最小單元間的相互作用的,是用量化等方法計算擬合後生成的經驗式,有人會嫌它粗糙,但是它確確實實給我們模擬大系統提供了可能,只能說關注的切入點不同罷了。常見的有三類力場:全原子力場,聯合力場,粗粒化力場;當然還有所謂第一代,第二代,第三代力場的說法,這裡就不一一列舉了。
再次提醒注意:必須選擇適合於我們所關注體系和我們所感興趣的性質及現象的力場。
3)通過實驗數據或者是某些工具得到體系內的每一個分子的初始結構坐標文件,之後,我們需要按我們的想法把這些分子按照一定的規則或是隨機的排列在一起,從而得到整個系統的初始結構,這也是我們模擬的輸入文件。
4)結構輸入文件得到了,我們還需要力場參數輸入文件,也就是針對我們系統的力場文件,這通常由所選用的力場決定,比如鍵參數和非鍵參數等勢能函數的輸入參數。
分子動力學
5)體系的大小通常由你所選用的box大小決定,我們必須對可行性與合理性做出評估,從而確定體系的大小,這依賴於具體的體系,這裡不細說了。6)由於初始構象可能會存在兩個原子挨的太近的情況(稱之為bad contact),所以需要在正式模擬開始的第一步進行體系能量最小化,比較常用的能量最小化有兩種,最速下降法和共軛梯度法,最速下降法是快速移除體系內應力的好方法,但是接近能量極小點時收斂比較慢,而共軛梯度法在能量極小點附近收斂相對效率高一些,所有我們一般做能量最小化都是在最速下降法優化完之後再用共軛梯度法優化,這樣做能有效的保證後續模擬的進行。
7)以平衡態模擬為例,你需要設置適當的模擬參數,並且保證這些參數設置和力場的產生相一致,舉個簡單的例子,gromos力場是用的范德華勢雙截斷來定范德華參數的,若你也用gromos力場的話也應該用雙截斷來處理范德華相互作用。常見的模擬思路是,先在NVT下約束住你的溶質(劑)做限制性模擬,這是一個升溫的過程,當溫度達到你的設定后, 接著做NPT模擬,此過程將調整體系的壓強進而使體系密度收斂。
經過一段時間的平衡模擬,在確定系統弛豫已經完全消除之後,就可以開始取數據了。如何判斷體系達到平衡,這個問題是比較技術性的問題,簡單的講可以通過以下幾種方式,一,看能量(勢能,動能和總能)是否收斂;二,看系統的壓強,密度等等是否收斂;三看系統的RMSD是否達到你能接受的範圍,等等。
8)運行足夠長時間的模擬以確定我們所感興趣的現象或是性質能夠被觀測到,並且務必確保此現象出現的可重複性。
9)數據拿到手后,很容易通過一些可視化軟體得到軌跡動畫,但這並不能拿來發文章。真正的工作才剛剛開始——分析數據,你所感興趣的現象或性質只是表面,隱含在它們之中的機理才是文章中的主題。
作用勢的選擇與動力學計算的關係極為密切,選擇不同的作用勢,體系的勢能面會有不同的形狀,動力學計算所
得的分子運動和分子內部運動的軌跡也會不同,進而影響到抽樣的結果和抽樣結果的勢能計算,在計算宏觀體積和微觀成分關係的時候主要採用剛球模型的二體勢,計算系統能量,熵等關係時早期多採用Lennard-Jones、morse勢等雙體勢模型,對於金屬計算,主要採用morse勢,但是由於通過實驗擬合的對勢容易導致柯西關係,與實驗不符,因此在後來的模擬中有人提出採用EAM等多體勢模型,或者採用第一性原理計算結果通過一定的物理方法來擬合二體勢函數。但是相對於二體勢模型,多體勢往往缺乏明確的表達式,參量很多,模擬收斂速度很慢,給應用帶來很大的困難,因此在一般應用中,通過第一性原理計算結果擬合勢函數的L-J,morse等勢模型的應用仍然非常廣泛。
分子動力學計算的基本思想是賦予分子體系初始運動狀態之後利用分子的自然運動在相空間中抽取樣本進行統計
分子動力學
但是通常情況下,體系各自由度中運動周期最短的是各個化學鍵的振動,而這種運動對計算某些宏觀性質並不產生影響,因此就產生了屏蔽分子內部振動或其他無關運動的約束動力學,約束動力學可以有效地增長分子動力學模擬的時間步長,提高搜索相空間的能力。
分子動力學可以用於NPT,NVE,NVT等系綜的計算,是一種基於牛頓力學確定論的熱力學計算方法,與蒙特卡洛
分子動力學
另外,在實際應用中,經常把分子動力學方法和蒙特卡羅法聯合使用。
分子動力學計算機模擬是研究複雜的凝聚態系統的有力工具。這一技術既能得到原子的運動軌跡,還能象做實驗一樣作各種觀察。對於平衡系統,可以在一個分子動力學觀察時間(observationtime)內作時間平均來計算一個物理量的統計平均值,對於一個非平衡系統過程,只要發生在一個分子
分子動力學
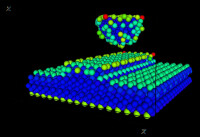
分子動力學
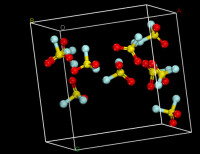
分子動力學
另外經驗勢近似有一個本身的局限性,它丟失了局域電子結構之間存在著的強相關作用信息,也就是說,不能得到成鍵性質(bondingproperties),以及原子動力學過程中的電子性質。儘管在某些特殊情況下,電子性質也能通過經驗勢近似來得到,例如用經驗勢的分子動力學來計算原子結構時,選取某幾種原子構型來作電子結構計算,這樣不僅耗時,而且原子動力學和電子結構計算成了相互獨立的過程。1985年,Car和Parrinello在傳統的分子動力學中引入了電子的虛擬動力學,把電子和核的自由度作統一的考慮,首次把密度泛函理論與分子動力學有機地結合起來,提出了從頭計算分子動力學方法(也稱CP方法),使基於局域密度泛函理論的第一原理計算直接用於統計力學模擬成為可能,極大地擴展了計算機模擬實驗的廣度和深度。
分子動力學模擬是研究微觀世界的有效手段"其勢函數和數值演演算法對模擬的精度有較大影響,為了提高勢函數的精確性,將基於局部密度泛涵理論的從頭計算分子動力學,量子化學分析參數擬合和蒙特卡洛方法相結合有望成為研究勢函數的最佳方法,隨著計算機性能的不斷提高,擺脫了經驗勢函數的從頭計算分子動力學的應用範圍將會不斷擴大,計算的精度也會不斷提高。所以,從頭計算分子動力學將會成為分子動力學模擬未來的主要發展方向。
數值演演算法的高速和高效也是人們一直奮鬥的目標,最近有人提出的多重時間寬度法,由於有效地減少了計算時間而可能成為分子動力學方法中較有前途的數值積分演演算法,分子動力學方法與其他計算方法,如有限單元法、模擬淬火法、蒙特卡羅法等的結合也將成為未來的發展方向之一。
詞條圖冊
計算化學分子模擬en:moleculardynamicsja:分子動力學法nl:Moleculairedynamica
http://wiki.keyin.cn/index.php/%E5%88%86%E5%AD%90%E5%8A%A8%E5%8A%9B%E5%AD%A6
基本信息
- 中文名
- 分子動力學
- 外文名
- Molecular Dynamics——MD
- 1957年
- 基於剛球勢的分子動力學法
- 1977年
- 約束動力學方法(Rychaert等)
- 1964年
- 質點系-擴張(Rahman)1971年