藥物代謝動力學
藥物代謝動力學
藥物代謝動力學(pharmacokinetics)簡稱葯代動學或葯動學,主要研究機體對藥物的處置(Dispostion) 的動態變化。包括藥物在機體內的吸收、分佈、生化轉換(或稱代謝) 及排泄的過程,特別是血葯濃度隨時間變化的規律。藥物的代謝與人的年齡、性別、個體差異和遺傳因素等有關。
藥物代謝動力學
包括藥物消除動力學
一級消除動力學:單位時間內消除的藥量與血漿藥物濃度成正比,又叫恆比消除
零級消除動力學:單位時間內體內藥物按照恆定的量消除,又叫恆量消除
藥物代謝動力學的重要參數:
1、藥物清除半衰期(half life),是血漿藥物濃度下降一半所需要的時間。其長短可反映體內藥物清除速度。
2、清除率(clearance,CL),是機體清除器官在單位時間內清除藥物的血漿容積,即單位時間內有多少毫升的血漿中所含藥物被機體清除。
3、表觀分佈容積(apparent volume of distribution),是當血漿和組織內藥物分佈達到平衡后,體內藥物按此時的血漿藥物濃度在體內分佈時所需的體液容積。
4、生物利用度(bioavailability),即經任何給葯途徑給予一定劑量的藥物後到達全身血液循環內藥物的百分比。
細胞膜和亞細胞膜(線粒體膜、微粒體、細胞核膜、小囊泡膜)總稱為生物膜。生物膜主要由蛋白質(60-75%)與不連續的脂質雙分子層(25-40%,主要是磷脂)所組成。蛋白質分佈在脂質層的兩側,有些則嵌入膜內部。膜上有膜孔(直徑約8Å)及特殊轉運系統。由於生物膜主要由脂質構成,故脂溶性藥物易通過;由於具有膜孔,所以水及水溶性、非極性小分子藥物也能通過;由於有特殊的轉運系統,所以水溶性大分子物質也能選擇性地通過生物膜。
藥物吸收、分佈、代謝和排泄過程中,藥物分子要通過各種單層(如小腸上皮細胞)或多層(如皮膚)細胞膜。儘管各種細胞結構不盡相同,但其細胞膜是藥物在體內運轉的基本屏障,藥物的通過方式和影響因素相似。
(一)被動轉運(簡單擴散和濾過):
藥物經膜孔擴散和脂溶擴散通過生物膜。特點:由高濃度向低濃度擴散,直至膜兩側濃度相等(動態平衡);不需酶,不耗能;無飽和現象,也不受其它轉運物質抑制;多屬外源性物質的轉運方式。被動轉運包括膜孔擴散和脂溶擴散。膜孔擴散(濾過):膜孔直徑約8Å(埃,1Å=10米),直徑<8Å或分子量<100的水溶性小分子物質均易通過膜孔擴散。脂溶擴散:即非離子擴散,細胞膜具有類脂結構,脂溶性藥物可溶於類脂質透過細胞膜,藥物的脂溶性越大越易擴散。擴散速度取決於膜兩側藥物濃度梯度及藥物在膜內的溶解度。受藥物解離度的影響也很大。藥物離解成陰、陽離子后,極性增加,脂溶性下降,難穿透類脂質屏障。
(二)特殊轉運:主要包括主動轉運,易化擴散和膜泡運輸。主動轉運又名“上坡”轉運:特點:是一種載體轉運,靠酶促,耗能;可逆濃度梯度透過細胞膜;兩種藥物轉運機制相同時,可出現競爭性抑制;有飽和現象;多屬內源性代謝物質的轉運方式。易化擴散:是通過鑲嵌在細胞膜上的多肽蛋白質來進行的。藥物與膜蛋白外側亞單位(載體)結合后,引起該蛋白質構型改變,將藥物甩向內側,再由該蛋白質的內側亞單位通過構型變化,進一步把藥物甩入細胞內。與主動轉運不同之處是順濃度梯度,不需酶促,不耗能;所需載體在藥物濃度高時可被飽和,轉運系統可被某些物質抑制或競爭。細胞表面內陷,優質膜把環境物質包圍成小泡,再脫離膜質進入細胞內的過程,為內吞作用;細胞內的細胞質小泡同膜質融合,把其所含物質排除細胞,為外排作用。
(三)載體運轉
載體轉運是指轉運體在細胞膜的一側與藥物或生理性物質結合后,發生構型改變,在細胞膜的另一側將結合的內源性物質或藥物釋出。
特點是:①對轉運物質有選擇性;②載體轉運能力有限,故具飽和性;③結構相似的藥物或內源性物質可競爭同一載體而具有競爭性,並可發生競爭性抑制。載體轉運主要發生在腎小管、膽道、血腦屏障和胃腸道的藥物轉運。
三、藥物的體內過程:即藥物被吸收進入機體到最後被機體排出的全部歷程,包括吸收、分佈、代謝和排泄等過程。其中吸收、分佈和排泄屬物理變化稱為轉運。代謝屬於化學變化亦稱轉化。機體對藥物作用的過程,表現為體內藥物濃度隨時間變化的規律。藥物動力學是研究藥物體內過程規律,特別是研究血葯濃度隨時間而變化的規律。
(一)吸收:藥物從給葯部位進入血循環稱為吸收。影響吸收的因素主要有: 1、給葯途徑:吸收速度:吸入>舌下>肌注>皮下>直腸>口服>皮膚。 2、藥物性質: (1)脂溶性:脂溶性越大,吸收越快; (2)水溶性:易溶於水的藥物易吸收; (3)離解度:不解離部分脂溶性較大,易吸收;而解離部分,由於帶有極性,脂溶性低,難以吸收。。口服藥物被吸收進入體循環的比率,即給藥量與吸收量的比率稱為生物利用度(或生物可用度)。
(二)分佈:影響藥物分佈的主要因素有: 1、藥物的性質:脂溶性大分佈到組織器官的速度快。 2、藥物與組織的親和力:有些藥物對某些組織器官有特殊的親和力。藥物對組織器官的親和力與療效及不良反應有關。 3、藥物與血漿蛋白(主要是白蛋白)結合率:結合率大小與療效有關。結合后: (1)無活性; (2)不易透過毛細血管壁,影響分佈和作用; (3)結合型藥物分子量大,不易從腎小球濾過,也不受生物轉化的影響;因此在體內的作用時間也延長。 4、血流量大小:腦、心肝、腎等組織器官血管豐富,血流量大,藥物濃度較高,有利於發揮作用,也易引起這些組織器官損害。 5、特殊屏障:血腦屏障是血液與腦組織之間的屏障,極性小而脂溶性大的藥物較易通過,對極性大而脂溶性小的藥物則難以通過。
(三)代謝(生物轉化)藥物代謝的主要器官是肝臟。也可發生在血漿、腎、肺、腸及胎盤。1、藥物代謝(轉化)酶:(1)肝微粒體葯酶:藥物在體內主要靠肝細胞微粒體的葯酶。其中最主要的是混合功能氧化酶系,其由三部分組成:血紅蛋白類,包括細胞色素P-450及細胞色素b5;黃素蛋白類,包括還原型輔酶Ⅱ-細胞色素C還原酶(或稱還原型輔酶Ⅱ-細胞色素P-450還原酶)及還原型輔酶I-細胞色素b5還原酶,是電子傳遞的載體;脂類,主要是磷脂醯膽鹼,功能尚不清楚。此三部分共同構成電子傳遞體系,使用使藥物氧化,三者缺一,藥物代謝就不能完成。 (2)細胞漿酶系:包括醇脫氫酶、醛氧化酶、黃嘌呤氧化酶等。一些藥物經微粒體葯酶氧化生成醇或醛后,再繼續由醇脫氫酶和醛氧化酶代謝。 (3)線粒體酶:包括單胺氧化酶、脂環族芳香化酶等。單胺氧化酶能使各種內源性單胺類(多巴胺、腎上腺素、去甲腎上腺素、5-羥色胺等)和外源性的胺類(乳酪或酵母中的酪胺等)氧化脫氨生成醛,再進一步氧化滅活。 (4)血漿酶系:包括單胺氧化酶、兒茶酚胺氧位甲基轉移酶、醯胺酶及假膽鹼酯酶等。前二者可氧化血漿中內源性或外源性單胺類物質。 (5)腸道菌叢酶系:能將某些營養物質變為胺類、羧酸或烴類等有毒物質,腸道菌大量繁殖,產胺過多,可能誘發嚴重肝功不良者的昏迷,故臨床上口服新黴素的目的是殺滅腸道菌叢減少胺類生成,從而減輕肝昏迷。2、代謝(轉化)類型:可分兩類。第一類包括氧化、還原及水解過程;第二類為結合過程,第一類轉化產物再經與體內某些代謝物結合,產物一般水溶性加大,利於排泄。 (1)第一階段反應(第一類型):氧化、還原及水解等。氧化,如醇氧化、醛氧化、單胺氧化、氧化脫氫及N-氧化等;還原,如硝基還原成氨基(-NH2)。 (2)第二階段反應(第二類型):即結合反應,使葯失效,隨尿排出。含羥基、羧基、胺基的化合物與葡萄糖醛酸結合成酯、醚、醯胺化合物;硫酸可與酚類藥物及酚性類固醇結合成硫酸酯;N-甲轉移酶使伯胺、腫胺及叔胺甲基化,以S-腺苷甲硫氨酸作為甲基供應體;磺胺類及芳香族氨基等在乙醯輔酶A參與下乙醯化。3、藥物代謝的意義:(1)解毒,絕大多數藥物通過代謝后失去藥理活性,稱為解毒。肝葯酶活性低時,應用主要在肝滅活的藥物時要特別慎重。 (2)活化,少數藥物經代謝變化後效力反而增強,稱為活化。4、葯酶的誘導劑和抑製劑:某些藥物可促進葯酶對其的降解,又可促進其它藥物的葯酶的降解作用,長期服用可產生耐受性。有些藥物能抑製藥酶的活性,從而延緩藥物的降解,長期應用可產生積蓄中毒。
(四)排泄:主要通過腎臟。此外還有肺、膽汁、乳汁、唾液腺、支氣管腺、汗腺、腸道等。1、腎臟排泄包括腎小球濾過和腎小管排泌。腎小球濾孔約600Å,分子量<65000均可通過。腎小管排泌是主動轉運過程,需要載體,腎小管上皮細胞具有兩類轉運系統(兩種載體):有機酸轉運系統,轉運有機酸藥物;有機鹼轉運系統,轉運有機鹼藥物。有飽和現象,對同一轉運系統有競爭性抑制。腎小管上皮細胞膜也具類脂結構,藥物可通過脂溶擴散從腎小管重吸收回到血液中去,腎小管重吸收的主要是未離解的脂溶性藥物,改變尿液pH可影響藥物的離解度,能顯著影響弱酸性或弱鹼性藥物在腎小管的重吸收;相反,增加弱酸性藥物的離解度,可減少其在腎小管的重吸收,加速其排泄率。故弱酸性藥物中毒時,宜用碳酸氫鈉鹼化尿液,加速毒物排出。腎功能不全者慎用或禁用主要經腎排泄的藥物。2、從膽汁排泄的藥物,除需具有一定的化學結構外,分子量要超過300才可以。分子量超過5000的大分子或蛋白質很難從膽汁排出。藥物從肝細胞向膽汁的轉運是主動轉運過程,需有載體,有飽和現象。肝細胞至少有三個轉運系統:有機酸類轉運、有機鹼類轉運和中性化合物轉運。屬同一轉運系統的藥物,有競爭性抑制。藥物由膽汁排入十二指腸后,有些從糞便排出,有些可被腸上皮細胞吸收入血液,形成“肝-腸循環”。 3、某些藥物可從乳汁排泄,可能引起乳兒中毒。4、某些揮發性藥物可從肺排泄。5、有些藥物可從支氣管排泄。6、有些可從汗腺排泄。
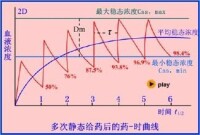
(一)、藥物濃度―時間曲線:給葯后藥物濃度隨時間遷移發生變化為縱坐標,以時間為橫坐標繪製曲線圖,稱為藥物濃度―時間曲線(見圖)。由於血液是藥物及其代謝物在體內吸收、分佈代謝和排泄的媒介,各種體液和組織中的藥物濃度與血液中的藥物濃度保持一定的比例關係,而有些體液採集較困難,所以血葯濃度變化最具有代表性,是最常用的樣本,其次是尿液和唾液。 (二)、消除速率類型: 1、一級速率消除:單位時間內體內藥物濃度按恆定比例消除。計算公式為:dC/dt ﹦―KC 2、零級速率消除:單位時間內體內藥物濃度按恆定的量消除。計算公式為:dC/dt ﹦―KoC° dC/dt ﹦―Ko 3、混合速率消除:少部分藥物小劑量時以一級速率轉運,而在大劑量時以零級速率轉運。因此描述這類藥物的消除速率需要將兩種速率類型結合起來,通常以米﹣曼氏方程式描述。 (三)、葯動學模型:房室模型是葯動學研究中廣為採用的模型之一,由一個或數個房室組成,一個是中央室,其餘是周邊室。這種模型是一種抽象的表達方式,並非指機體中的某一個器官或組織。 (四)、葯動學參數計算及意義: 1、峰濃度和達峰時間:指血管外給葯后藥物在血漿中的最高濃度值及其出現時間,分別代表藥物吸收的程度和速度。 2、曲線下面積:指時量曲線和橫坐標圍成的區域,表示一段時間內藥物在血漿中的相對累積量。 3、生物利用度:藥物經血管外給葯后能被吸收進入體循環的分量及速度。 4、生物等效性:比較同一種藥物的相同或者不同劑型,在相同試驗條件下,其活性成分吸收程度和速度是否接近或等同。 5、表觀分佈容積:指理論上藥物均有分佈應佔有的體液容積。 6、消除速率常數:指單位時間內消除藥物的分數。 7、半衰期:指血漿中藥物濃度下降一半所需要的時間。 8、清除率:指單位時間內多數毫升血漿中的藥物被清除。
合理的給藥方案是使穩態血漿藥物濃度(Css)達到一個有效而不產生毒性反應的治療濃度範圍,稱為靶濃度(target concentration)。
在大多數情況下,臨床多採用多次間歇給葯或是持續靜脈滴注,以使穩態血漿藥物濃度維持在靶濃度。因此要計算藥物維持劑量(maintenance dose)
t1/2才能達到穩態血葯濃度,增加劑量或者縮短給葯間隔時間均不能提前達到穩態,只能提高藥物濃度,因此如果患者急需達到穩態血葯濃度以迅速控制病情時,可用負荷量(loadingdose)給葯法。負荷量是指首次劑量加大,然後再給予維持劑量,使穩態血葯濃度(即事先為該患者設定的靶濃度)提前產生。

基本信息
- 中文名
- 藥物代謝動力學
- 外文名
- pharmacokinetics
- 別名
- 葯代動學或葯動學
- 方法
- 數學原理和方法