間變性大細胞淋巴瘤
間變性大細胞淋巴瘤
間變性大細胞淋巴瘤(anaplastic large cell lymphoma , ALCL) 亦稱 ki-1 淋巴瘤,細胞形態待殊,類似 R-S 細胞,有時可與霍奇金淋巴瘤和惡性組織細胞病混淆。細胞呈 CD30 + ,亦即 Ki-1(+), 常有 t (2 ; 5) 染色體異常,臨床常有皮膚侵犯,伴或不伴淋巴結及其他結外部位病變。免疫表型可為 T 細胞型。臨床發展迅速,治療同大細胞淋巴瘤。
間變性大細胞淋巴瘤,即是非霍奇金淋巴瘤的一種獨立類型,由德國病理學家Stein等於1985年應用Ki-1(CD30)抗體識別,常呈間變性特徵,被命名為間變性大細胞淋巴瘤。REAL分類將B細胞表型者歸為瀰漫性大B細胞性淋巴瘤。目前,ALCL只包括T表型和Null(非T非B)表型。約60%-85%左右ALCL病例表達間變性淋巴瘤激酶(anaplasticlymphomakinase,ALK)融合蛋白,這是由於2號染色體上的ALK基因位點的畸變所致。最常見的是t(2;5)(p23;q35)而形成融合基因NPM-ALK,它是由位於5號染色體上的核仁磷酸蛋白B23(NPM)基因與位於2號染色體的ALK基因相融合形成,表達融合蛋白為NPM-ALK蛋白;最近尚有更多的ALK基因與其他基因通過染色體轉位或者是染色體的倒轉而形成的融合基因被發現,如t(1;2)(q25;p23)所形成的TPM3-ALK基因,t(2;3)(p23;q21)產生的TFG-ALKs基因,TFG-ALKL基因和TFG-ALKxL基因,inv(2)(p23;q35)所形成的ATIC-ALK基因,t(2;17)(p23;q23)形成的CLTCL-ALK基因及t(X;2)(q11;p23)形成的MSN-ALK基因。
在臨床上ALCL被分為原發性(系統性和皮膚)及繼發性(由其他淋巴瘤轉化而來)兩種,約佔全部NHL的2%-7%。由於越來越多研究表明原發性系統性ALCL中ALK陽性和ALK陰性病例其表現有明顯差異,因此將ALK陽性和ALK陰性的原發性系統性ALCL分別介紹。
ALK陽性的原發性系統性ALCL主要發生在30歲之前的病人。Falini等的研究還表明其性別差異很明顯,男女比率為6:1,並且主要發生在20-30年齡段。ALCL通常表現為外周和腹部淋巴結的腫大。約有2/3的病人有發熱或者是III/Ⅳ期。在約60%的病例有結外的累及,約40%有兩個或兩個以上結外被累及。而皮膚(21%)、骨(17%)和軟組織(7%)是最常見的被累及的結外部位。眾多的研究表明ALK陽性的ALCL其預后明顯好於ALK陰性的病例。
ALK陰性的原發性系統性ALCL與ALK陽性的病例有許多形態學、免疫表型及臨床特徵都是相同的,ALK融合基因的檢測是區別他們的唯一方法。ALK陰性原發性系統性ALCL更多見於年齡大的病人,且這些病人的預后較差。
原發性皮膚性ALCL約佔皮膚淋巴瘤的10%左右。目前已證實它和ALK陰性原發性系統性ALCL是兩個不同的實體。原發性皮膚性ALCL多發生在老年病人,平均年齡約為60歲左右,ALK陰性且缺乏細胞毒性表型。病變表現為實體的、無癥狀的皮膚或皮下紫紅色腫塊。表面可發生潰瘍,較少見的是多腫瘤結節的形式侵犯周邊區域或多部位、多中心發生腫瘤為特徵。在約25%的病人可有部分或全部消退,而經局部切除並加以化療具有良好的預后。
淋巴結結構部分或全部破壞,有的只侵犯淋巴竇,瘤細胞常首先累及淋巴結副皮質區,然後成巢狀或沿淋巴竇彌散性播散。淋巴結結構部分破壞時,瘤細胞常侵犯副皮質區和濾泡旁,血管浸潤較明顯,常見纖維組織增生,表現為包膜增厚,纖維條索包繞瘤細胞巢。增生的瘤細胞可呈單一性或伴有其他成分如小淋巴細胞、漿細胞、中性粒細胞、組織細胞等,少數可見吞噬細胞和壞死。
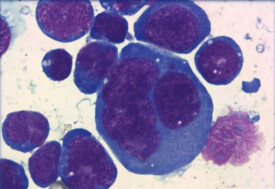
細胞體積較大或中等,呈圓形,橢圓或不規則。核為圓形、卵圓形或不規則形,有胚胎樣核,其核形彎曲,核膜一側平滑微凸,另一側凹陷有多個切跡。有的瘤細胞核類似霍奇金的R-S細胞樣的雙核瘤細胞,但無診斷性R-S細胞。有時可見排列為馬蹄形或花環狀的多核巨細胞,染色質為粗塊狀,核仁明顯嗜酸性。
瘤細胞形態多樣,核不規則,偏位,核仁大,胞質豐富。胞質中見大量堆積核糖體、粗面內質網。胞質內散在膜包被的高電子密度顆粒和透明的囊泡,其是否為穿孔素,顆粒酶,尚待證實。高爾基器位於核膜凹陷處。瘤細胞偶見原始細胞連接,但無橋粒。
根據瘤細胞主要形態學特徵分為3個亞型。
(70%)
病變淋巴結包膜增厚,全部或部分淋巴竇受累,瘤細胞沿淋巴竇,副皮質區浸潤,早期圍繞小血管生長。常見特徵性的胚胎樣、花環樣及R-S樣巨細胞。
(10%)
組織結構與普通型基本一致。瘤細胞體積小到中等,散在或呈小灶分佈。同時伴有大量的組織細胞。CD30陽性瘤細胞散在分佈。
(smallcellvariant5%-10%)
淋巴結結構部分或完全破壞,瘤細胞體積較小,核形不規則,部分呈腦回狀,染色質緻密。瘤細胞間可見散在或呈簇狀分佈的無明顯異型性的大細胞。CD30陽性的瘤細胞特徵性的聚集在高內皮靜脈周圍。
需要指出的是,上述類型偶可在同一個活檢病變中共存或在同一個病例先後不同活檢病變中出現,說明ALCL具有廣泛的形態學譜。
對ALCL確切的病理機制尚知之不多。研究發現,約有30-80%患者存在2號染色體間變性淋巴瘤激酶(anaplasticlymphomakinase,ALK)基因易位,產生有致癌性的異常ALK融合蛋白。ALK陽性ALCL中最常見的核型異常是t(2;5)(p23;q35)易位,即ALK基因與5號染色體的核磷酸蛋白(nucleophosmin,NPM)基因融合而表達NPM-ALK融合蛋白,由於野生型NPM部分含有核定位位點,NPM-ALK融合蛋白可以進入核內,該核型異常約佔到ALK陽性ALCL的75%。最近尚有更多的ALK基因與其他基因通過染色體轉位或者是染色體的倒轉而形成的融合基因被發現,如t(1;2)(q25;p23)所形成的TPM3-ALK基因,t(2;3)(p23;q21)產生的TFG-ALKs基因,TFG-ALKL基因和TFG-ALKxL基因,inv(2)(p23;q35)所形成的ATIC-ALK基因,t(2;17)(p23;q23)形成的CLTCL-ALK基因及t(X;2)(q11;p23)形成的MSN-ALK基因。
受體型酪氨酸激酶間變性淋巴瘤激酶(Anaplastic lymphoma kinase,ALK)最早發現於間變性大細胞淋巴瘤(Anaplastic large cell lymphoma,ALCL)中,由2號及5號染色體易位所形成的融合蛋白質包含了ALK的3’端胞內結構域,以及核磷蛋白(Nucleophosmin,NPM)的5’端的結構域。隨後的研究發現,正常的ALK專一表達於神經系統中,如腦和神經索,尤其是新生兒的腦中。
ALK基因位於染色體2p23位點,正常情況下人源的alk可轉錄產生大小6222bp的mRNA,由29個外顯子構成,編碼1620個氨基酸序列200KDa的I型穿膜蛋白ALK,該蛋白為一種受體酪氨酸激酶(receptortyrosinekinase,RTK),是RTK胰島素超家族的成員。完整的ALK具有典型的RTK三部分結構,即胞外區、親脂性穿膜區和胞漿內酪氨酸激酶。據文獻報道,ALK蛋白除在極少部分瀰漫性大B細胞淋巴瘤中表達外,可在60%~85%的原發性系統性ALCL中表達,是原發性系統性ALCL相對特異的免疫表型特徵。
ALK在某些ALCL中的異常表達來源於不同的染色體易位。ALK易位的基因組斷裂點多發生在16及17號外顯子中間的內含子,而17-26號外顯子編碼ALK胞內結構域,每個易位產生一種不同的融合蛋白質,由配偶體的5’端和ALK酪氨酸激酶結構域3’端融合得到。大多數情況下,5’端的配偶體具有可以形成同源或異源二聚體的結構域,使得ALK激酶結構域交互磷酸化,相互作用增強並且使多種下游蛋白質磷酸化。失去調控的ALK活性增高,使其功能近似原癌蛋白質,這些融合蛋白質定位在不同的亞細胞區域上,因此可能導致不同的細胞功能改變。
大約70-80%的ALK陽性的ALCL表達NPM-ALK,它是由染色體t(2;5)(p23;q35)易位引起的,npm的5’端與alk的3’端融合,導致NPM的氨基端與ALK梭基端的酪氨酸激酶功能區融合。位於5號染色體的npm編碼一種調節細胞周期的NPM,該蛋白的分子量為38kd,與前核糖體顆粒運輸及核糖體生物發生,調節細胞分裂、DNA修復、轉錄和基因組穩定性相關。NPM包含核定位信號以及二聚體結構域,可以產生大的同源二聚體及異源二聚體。NPM對NPM-ALK融合蛋白髮揮轉化功能非常重要,缺乏NPM二聚體結構域的突變體小能轉化細胞,提示二聚化作用是信號傳遞的關鍵因素。轉基因模型小鼠的研究結果顯示NPM-ALK可導致惡性的淋巴瘤。
正常情況下ALK只在神經系統中表達。人體中ALK基因表達水平隨著腦的發育成熟而下降,成熟腦組織中的量很低,表達存在一定的區域性;其它系統尤其是造血系統中未發現ALK的表達。ALK基因在絕大多數非造血系統腫瘤和正常組織中缺乏表達,表明ALK蛋白的分佈範圍是極其狹窄的。ALK蛋白是ALCL重要的分子標誌物,在ALCL的診斷中具有很高的價值。
也稱B23,最早在70年代末及80年代初被鑒定。NPM是由5號染色體所編碼,分子量為38kD的核仁蛋白,NPM分子可通過其N端的一個寡聚區模板及C端(梭基端)的2個核定位信號與核蛋白結合,參與了細胞質/核運輸及細胞內核糖體前顆粒的裝配核運輸。NPM不停地穿梭於核仁與胞漿之間,因而可作為一種載體將新合成的蛋白質運轉至核仁。NPM帶有寡聚功能的結構域,正常情況下會發生自身的寡聚,也可以與NPM-ALK形成異聚體,會導致NPM-ALK蛋白在核內的聚集。
NPM-ALK:t(2;5)(p23;q35)染色體重排導致了一種80KD的融合蛋白的表達,它包括了NPM的前117aa融合到了ALK的C末端1058-1620殘基。ALK基因組序列的斷點是在1935bp內含子處,該內含子位於編碼ALK跨膜區和近膜區域的外顯子之間。NPM基因序列的斷點位於NPM的內含子4處。幾乎所有包括了ALK的融合蛋白中均包含了相同的構成ALK細胞質部分的563aa,僅MSN-ALK由於斷點位於ALK基因的近膜區外顯子處而ALK部分要較其他稍短,但是結構也非常相似。
ALK分子胞漿內的尾端攜帶有酪氨酸激酶分解區,t(2;5)使編碼NPM的N端結構域的基因部分與編碼全部ALK蛋白胞漿部分的基因位置靠近,結果使ALK基因受到NPM啟動子的調控,從而誘導NPM-ALK融合基因完全的、普遍的轉錄,產生名為NPM-ALK或P80的80kd的融合蛋白,NPM-ALK在ALCL中是最常見的一種ALK融合蛋白,也是目前研究最多、淋巴瘤的預后最好。
ALCL是一種高度惡性淋巴瘤,5年生存率為52%。治療可實施放療、化療、骨髓移植等方法。化療最為適宜,多數病例可完全緩解(CR),複發率低,3年和5年生存率均較高。放療起初效果良好,但遠期易複發。骨髓移植被認為是一種有效的應急治療措施。一般說來,ALCL比其它大細胞淋巴瘤的預后好。其預后與腫瘤的發生年齡,有無癥狀,原發部位,臨床分期及免疫學分型有關,與組織學分型無明顯關係。兒童及青年對治療反應好,5年生存率遠高於成年人。臨床無癥狀者較有癥狀者預后好,原髮結內較結外者預后好。臨床I,II期病例較III,IV期病例3年生存率高,而組織學分型與預后無關。原發於皮膚ALCL,特別是局限於皮膚者,因具有良好的預后(4年生存率達80%),且部分病例能自愈(17%)以及病灶行單純切除或局部放療的療效與治療高惡淋巴瘤的系統多重化療或自身骨髓移植的療效相同,而被認為是一種獨特類型的低度惡性腫瘤。
許多學者認為ALCL患者預后與其染色體是否發生易位有關。在兒童和年青患者的侵襲性NHL中,ALK陽性系統性ALCL治癒可能性最大,預后優於任何其他形式的外周T細胞淋巴瘤。ALK陽性和ALK陰性ALCL之間預后的差異性首先由Shiota等報道,他們指出,ALK陽性病例的5年生存率(79.8%)遠好過ALK陰性病例(32.9%),P<0.01。Brunangelo Falini等對78例ALCL(ALK陽性53,ALK陰性25)患者預后統計顯示,ALK陽性病例中有77.3%獲得完全緩解,15.0%獲得了部分緩解,緩解率達到92.3%,同時4例(7.7%)對化療耐受;ALK陰性病例中有56.0%獲得完全緩解,28.0%獲得了部分緩解,緩解率達到84.0%,同時4例(16.0%)對化療耐受;所有病例中位生存年領為2.1年(0.07年-13.17年),ALK陽性病例總生存率(71.0%±6.0%)遠好過ALK陰性病例(15.0%±11.0%),P<0.0007。 ALK陽性病例10年無病生存率(82.0%±6.0%)也遠好過ALK陰性病例(28.0%±14.0%),P<0.0001。Randy D等研究也顯示ALK陽性ALCL病例的5年生存率(93.0%)遠好過ALK陰性ALCL病例(37.0%),P<0.00001。眾多研究表明有ALK融合基因的ALCL病例的預后明顯好於陰性病例,但確切的機制倘不清楚,Brunangelo Falini等研究顯示ALK陽性病例對化療耐受(7.7%)明顯低於ALK陰性病例(16.0%),同時ALK陽性病例化療效果明顯好過ALK陰性病例,儘管確切的機制不清楚,但有一點是可以肯定的,就是ALK陽性的ALCL的預后明顯好於ALK陰性病例,因此對於臨床診斷來說很有必要檢測ALCL中有無ALK融合基因。目前,染色體易位和ALK的表達已經被WHO規定為ALCL的臨床診斷指標之一。
基本信息
- 中文名
- 間變性大細胞淋巴瘤