早衰症
遺傳病
早衰症(兒童早老症)屬遺傳病,身體衰老的過程較正常快5至10倍,患者樣貌像老人,器官亦很快衰退,造成生理機能下降。病徵包括身材瘦小、脫髮和較晚長牙。患病兒童一般只能活到7至20歲,大部分都會死於衰老疾病,如心血管病,現未有有效的治療方法,只靠藥物針對治療。
早衰症(Hutchinson-Gilford Syndrome),全稱早年衰老綜合症(Hutchinson-Gilford Progeria syndrome),又稱兒童早老症,屬遺傳病,Hutchinson於1886年首先報告。
雖然本病為一種先天遺傳性疾病,但還不能確定是常染色體隱性還是顯性遺傳。本病為一綜合征,特點為發育延遲,至嬰兒時期就發生進行性老年性退行性改變。患者身體的老化過程十分快速。而罹患此病孩童的年齡很少超過13歲,大約每八百萬個新生兒之中就有一位患有此疾。

早衰症患者
患者作為一名新生兒,出生時通常都很正常。然而,一年之內,他們的成長速度減緩,不久之後,他們個子比其它正常孩子要矮,體重要輕。雖然具備正常的智力,但頭髮斑禿、皮膚褶皺而鬆弛、牙齒脫落,鼻子扁縮,臉與下巴偏小,與腦袋大小極不相稱,眼睛凹陷,在面頰和手臂的皮膚上分佈著若隱若現的老年斑,整個的像一位80歲的老人。
他們還患有老年人才有的多種老年疾病,如關節僵硬、髖部脫臼和嚴重的心臟血管疾病。然而,與正常老年人有關的其它疾病如白內障和骨關節炎,在兒童早衰症患者身上就沒有出現。
患有兒童早衰症的兒童其身體衰老速度比正常衰老過程快5~10倍,使其貌如老人。患者體內的器官亦快速衰老,造成各種生理機能下降。早衰症病童較常出現的癥狀包括:脫髮、較晚長牙、身材矮小及皮下脂肪減少等,但病童的心智年齡大多與同齡兒童無異。專家指出,病童一般只能活到7~20歲,並大多死於心血管疾病等衰老病。目前沒有有效治療早衰症的方法。
早衰症的早期癥狀包括了發育遲緩、局部性硬皮病癥狀。當患者過了幼年期之後,其他的癥狀會變的更明顯。
獨特外觀:身材矮小,體重下降且和身高不成比例,性發育不成熟。皮下脂肪組織減少。頭和面不成比例,頭部所佔面積相對較大,而面部相對較小。下頜比正常人小。頭皮靜脈明顯,脫髮呈普遍性。眼呈鳥眼樣外形,牙齒髮育延遲,胸呈梨形,鎖骨短而發育不良,姿勢呈騎馬形,兩腳分開的寬度大,走路時拖著兩腳,髖外翻。大拇指細,關節永久性強直 最有特徵性的臨床表現為皮膚變薄、緊張、乾燥、皺摺。
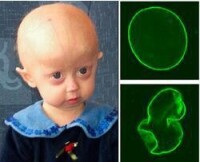
早衰症
這種患兒在嬰兒期常常是正常的,或僅有硬皮病樣癥狀。面中部青紫和鉤狀鼻常提示有本病的可能性。到1歲左右癥狀愈來愈明顯,直到第二年呈現各種特徵性表現。本病患者一般無甲狀腺、甲狀旁腺、垂體和腎上腺方面的異常。但基礎代謝率增加,血脂不正常。早老症患者可發生動脈粥樣硬化。由於心血管和腦血管的病變常早年夭折。“動脈硬化”是早衰症患者常見的癥狀。在早衰症患者中,高血脂的問題是由於低密度脂蛋白增加及血中膽固醇增加所造成的。而動脈硬化的問題在每個早老症患者身上皆可看見。根據此臨床發現,科學家推測和脂肪酸運輸及氧化相關的基因表現亦可能為導致早衰症的可能原因之一。
早衰症研究基金會的研究人員於2004年6月17日宣布,Lamin A基因的突變,是導致早衰症兒童細胞結構及功能逐漸退化的原因。早衰症的全名為Hutchinson-Gilford早衰症綜合症(HGPS或Progeria)。

早衰症
除此之外,早衰症患者通常對陽光敏感,這是因為此病患者的身體無法進行正常人的DNA修復工作,而失掉細胞複製及蛋白質製造的功能。目前,有兩個與早衰症有關的基因缺陷──CSA和CSB被發現,其中CSA基因位於第5號染色體上。這兩個基因密碼產生的蛋白質都與DNA修復以及複製轉譯機能有關。
“動脈硬化”是早衰症患者常見的癥狀。在早衰症患者中,高血脂的問題是由於低密度脂蛋白增加及血中膽固醇增加所造成的。而動脈硬化的問題在每個早老症患者身上皆可看見。根據此臨床發現,科學家推測和脂肪酸運輸及氧化相關的基因表現亦可能為導致早衰症的可能原因之一。不像其他加速性老化疾病,例如Werner綜合症、Cockayne綜合症或著色性干皮症,早衰症並非是由具有缺陷的DNA修復程序所引起。因為這些“加速老化疾病”展現了老化的不同面向,但是並非是每個面向。它們常被稱之為“部分早衰症”。
基因變異導致此怪病
2003年,美國國立人類基因組研究所(NHGRI) 、兒童早衰症研究基金會、紐約州學院和美國密歇根州大學的研究人員發現了何奇森-吉爾福德兒童早衰症是由單個基因的微小變異導致的。他們發現,兒童早衰症並非遺傳所至,而是人體內一種名叫LMNA(Lamin A)的蛋白基因發生突變導致。LMNA主要負責細胞核之間的相互鏈接,一但它發生突變,將會使細胞核處於不穩定狀態,加快人體的發育和衰老,其速度相當於正常速度的8倍,從而導致早衰。
研究小組負責人弗朗西斯·柯林斯博士:“我現在要宣布的是,這段獨立的基因(LMNA)現在能被複製成7種不同的基因,通常基因的突變結果取決於發生基因突變的位置。也就是說基因發生突變位置的不同,將會導致人們患上不同的疾病,比如肌肉營養失調、脂肪代謝障礙、原發性心肌病、以及神經病等疾病,所有這些都可能因為基因突變而產生,我們現在關注的這段引起兒童早衰症的基因十分特殊,因為它(LMNA)直接影響基因的鏈接,(它的突變)會對整個人體造成重大的影響。"

早衰症
兒童早衰症是一種十分罕見的兒童身體機能嚴重失調的疾病。究竟是什麼原因導致兒童早衰症的發生,一直以來這個問題長期困擾著醫學界,目前還沒有針對兒童早衰症的有效治療。兒童早衰症患者約翰·塔科特:“他們找到了我們的致病基因這個消息,的確令人激動,因為我們將有可能被治癒,死於心血管疾病或動脈硬化的兒童早衰症患者,其平均年齡通常在13周歲。兒童早衰症致病基因的發現,不僅有助於科學家儘早找到兒童早衰症的治療方法,還將有助於科學家深入研究人體衰老的過程和心血管疾病方面的諸多問題。
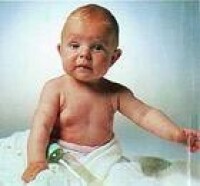
早衰症
早衰症最早是在1886年由喬納森·何奇森博士和黑斯廷斯·吉爾福德博士共同發現的。何奇森博士於1886年首次描述了這種病症,吉爾福德博士於1904年發現這一病症。
早衰症的診斷是根據患者的癥狀,患者的外觀和生長紀錄是相當重要的診斷依據。早老症孩童除了有生長遲緩的問題外,最常見的表徵如下:
典型的鳥型頭,且為禿頭;
身材矮小,體重不足;
四肢瘦且關節變得明顯;

早衰症
梨狀胸,且鎖骨短;
青筋突出。
實驗室檢查:一般沒有異常改變,有時可伴血清膽固醇增加和脂蛋白異常,餐后2h查血清甘油和脂肪酸含量也是正常的。實驗研究證明,脂肪組織中遊離脂肪酸的釋放度正常,尿中葡糖醛酸)的排泄也正常。Villee等認為本病的主要異常改變與某些結構蛋白質如膠原蛋白、肌動蛋白、肌凝蛋白和角蛋白的代謝和合成障礙有關。
臨床實驗室檢查:尿中玻尿酸值會增加,血中脂肪值常會不正常。但此些檢查對於診斷的幫助不大。
X光檢查:病患1~2歲時即會發現頭顱骨、胸部、長骨及指骨會發生變化,另外會有骨質疏鬆,和軟組織缺少的現象。
基因突變分析:早老症研究基金會已經發展出一套 "Diagnostic Testing Program" ,可透過此系統來了解基因是否已發生改變或突變。
早衰症的療法目前沒有一項被證實是有效的。大部分的治療集中在減少併發症,例如冠狀動脈繞道手術或低劑量阿司匹林。患者也可能受惠於高熱量飲食療法。
生長激素療法(Growth hormone treatment)也曾被嘗試。

早衰症
如有內分泌功能低下,應作相應的補充性治療;血脂高及動脈粥樣硬化表現者,應限制食物中的脂肪量,適當給予抗粥樣硬化的藥物。皮膚乾燥變硬者可內服煙酸、維生素E、維生素B族,口服丹參片或靜脈注射丹參液。有些發育畸形,可用外科手術進行矯正。總之,本病以對症處理為主,沒有特效治療方法。
據《每日科學》網站報道,一項最新研究指出,家庭小藥箱里常備的維生素C或許能治療可加速衰老的紊亂,尤其是成人早衰症(Werner's syndrome),這項研究成果發表在2010年1月的《美國實驗生物學會聯合會雜誌》上。
加拿大科研組在這份科研報告中指出,維生素C阻止甚至逆轉了患成人早衰症的老鼠的加速衰老過程,不過這項發現或許還適用於其它早衰症。患有成人早衰症的人,從20多歲開始表現出加速衰老的跡象,在50歲之前形成老年性疾病,他們一般會在50歲之前死亡。這篇論文的聯合作者,加拿大魁北克的邁克·勒貝爾說:“我們的研究顯然說明,沒患有疾病的健康生物或個體,不必通過服用大量維生素C來延長壽命,尤其當他們擁有均衡飲食,並經常進行鍛煉時。當生物或個體擁有WRN 基因變種,或者任何受WRN蛋白影響的基因時,他們傾向於患老年性疾病,這些人服用一定數量的維生素C會給健康帶來好處。”
科學家在健康老鼠和攜帶可引發成人早衰症(WRN基因)基因變種的老鼠的飲水裡加入了維生素C。在進行治療前,攜帶WRN基因變種的老鼠更易發胖、患糖尿病,並會慢慢形成心臟病和癌症。進行治療后,攜帶突變基因的老鼠變得跟正常老鼠一樣健康,壽命期限達到正常水平。維生素C還促進了老鼠儲存和燃燒脂肪的能力,減少了組織炎症,並降低了攜帶WRN基因的老鼠的氧化應激。健康老鼠沒表現出從維生素C受益的跡象。《美國實驗生物學會聯合會雜誌》的總編輯傑拉爾德·威斯曼說:“維生素C已經成為我們藥箱和食品中被人誤解最深的物質,這項研究和其他類似研究有助於解釋,這種化學物質是如何幫助一些人而不是所有人預防早衰,以及幫助他們預防早衰的原因。”
病例一:7歲的艾香蒂·艾略特·史密斯是英國布賴頓市附近人,當她在2003年5月出生時,體重2.55公斤的她看起來就像是一個正常而健康的嬰兒。然而當艾香蒂出生3周后,她的身體就突然開始抽筋,父母迅速將她送往醫院檢查,醫生為艾香蒂進行了一系列醫學測試,但並沒有查出她患有任何毛病。不過在接下來的幾個月中,艾香蒂的病情開始更加惡化、並變本加厲起來。直到艾香蒂一歲生日前,倫敦大奧蒙德街兒童醫院的專家們才診斷出艾香蒂患上了罕見的“哈欽森·吉爾福德早衰綜合征”。母親菲比回憶說:“我當場吃驚得暈厥了過去。當我蘇醒過來時,醫生告訴我,我們必須像照顧一名老奶奶一樣照顧艾香蒂。”據醫生稱,早衰症通常是由基因缺陷引發的,但並不一定會遺傳,艾香蒂的4歲妹妹布蘭迪·婁就沒有患上早衰症。兒童“早衰症”可說相當罕見,即使在全世界範圍內,目前已知也僅有52名早衰症患者,而艾香蒂是英國僅有的兩名早衰症患者之一。兒童早衰症患者出生時一般都表現正常,但到一歲左右就會加速衰老,並出現禿頂、關節炎、血管硬化、心臟疾病等各種老年病問題。

艾香蒂和母親
據英國醫學專家稱,到艾香蒂10歲時,她的身體將會衰老得像一名8旬老嫗。然而,勇敢的艾香蒂卻一直不屈不撓地和早衰症做鬥爭,她甚至還像其他同齡孩子一樣到一家主流學校去上學。不過,由於早衰症患者的平均壽命最多只有13歲,所以艾香蒂的生命已經開始進入“倒計時”,她的父母甚至放棄工作專門在家陪伴和照顧女兒,並將和女兒相處的每一個日子都當作是上天的恩賜。
病例二:現年18歲的英國女孩艾米·休斯和17歲的美國男孩尼克·賈米尼特是一對少年情侶。讓人心碎的是,兩人都患有一種罕見的早衰症——科凱恩綜合征,雖然他們都不到20歲,但他們的身體都已經如同六、七十歲的老人。他倆可能都無法活過20歲!不過,雖然他們無法一起慢慢變老,但他們已經發誓生死相依,尼克已在去年夏天正式向艾米求婚,他們計劃在今年夏天舉行一場浪漫的婚禮,在今生結為夫妻。不過據醫學專家稱,他們甚至可能無法活到今年夏天參加他們的夢幻婚禮!
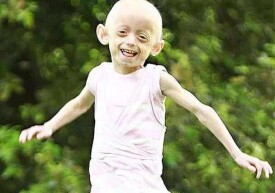
基本信息
- 中文名
- 早衰症
- 外文名
- Hutchinson-Gilford Syndrome
- 別名
- 兒童早老症、早年衰老綜合症
- 季節分佈
- 四季
- 是否傳染病
- 否
- 癥狀表現
- 兒童其身體衰老速度比正常衰老過程快5~10倍,貌如老人;體內的器官亦快速衰老,造成各種生理機能下降。
- 就診科室
- 內科