共找到2條詞條名為毛細管電泳的結果 展開
- 毛細管電泳
- 高效毛細管電泳法
毛細管電泳
毛細管電泳
毛細管電泳(capillary electrophoresis,CE)又稱高效毛細管電泳(high performance capillary electrophoresis,HPCE),是一類以毛細管為分離通道、以高壓直流電場為驅動力的新型液相分離技術。毛細管電泳實際上包含電泳、色譜及其交叉內容,它使分析化學得以從微升水平進入納升水平,並使單細胞分析,乃至單分子分析成為可能。長期困擾我們的生物大分子如蛋白質的分離分析也因此有了新的轉機。
毛細管電泳
1 雙電層
雙電層是指兩相之間的分離表面由相對固定和遊離的兩部分離子組成的與表面異號的離子層,凡是浸沒在液體中的界面都會產生雙電層。在毛細管電泳中,無論是帶電粒子的表面還是毛細管管壁的表面都有雙電層。
2 Zeta 電勢

毛細管電泳
石英材質的毛細管是毛細管電泳中最常使用的毛細管,管子內表面在pH>3 情況下帶負電,當其與溶液接觸時,會形成緊貼內表面的和遊離的兩部分離子。這兩部分離子組成的與表面電荷異號的離子層,即為雙電層,其中第一部分又稱之為Stern 層。第二層為擴散層。擴散層中遊離部分離子的電荷密度隨著和表面距離的增大而急劇減小。在Stern 層和雙電層的遊離部分的起點的邊界層之間的電勢稱之為管壁的Zeta 電勢。典型值大體在0-100 mV 之間,Zeta 電勢的值隨距離增大按指數衰減,使其衰減一個指數單位所需的距離稱之為雙電層的厚度(δ)。熔硅表面的Zeta 電勢與它表面上的電荷數及雙電層厚度有關,而這些又受到離子的性質、緩衝溶液pH 值、緩衝溶液中陽離子和熔硅表面間的平衡等因素的影響。
3 淌度、絕對淌度和有效淌度
帶電粒子在直流電場作用下於一定介質(溶劑)中所發生的定向運動稱為電泳。單位電場下的電泳速度稱為淌度。在無限稀釋溶液中(稀溶液數據外推)測得的淌度稱為絕對淌度。
電場中帶電離子運動除了受到電場力的作用外,還會受到溶劑阻力的作用。一定時間后,兩種力的作用就會達到平衡,此時離子作勻速運動,電泳進入穩態。實際溶液的活度不同,特別是酸鹼度的不同,所以樣品分子的離解度不同,電荷也將發生變化,這時的淌度可稱為有效電泳淌度。一般來說,離子所帶電荷越多、離解度越大、體積越小,電泳速度就越快。
4 電滲、電滲流和表觀淌度
電滲是推動樣品遷移的另一種重要動力。所謂電滲是指毛細管中的溶劑因軸向直流電場作用而發生的定向流動。電滲是由定域電荷引起。定域電荷是指牢固結合在管壁上、在電場作用下不能遷移的離子或帶電基團。在定域電荷對溶液中的反號離子吸引下形成了所謂的雙電層,致使溶劑在電場作用(以及碰撞作用)下整體定向移動而形成電滲流(毛細管中的電滲流為平頭塞狀)。
毛細管區帶電泳條件下,電滲流從陽極流向陰極。電滲流大小受到Zeta電勢、雙電層厚度和介質粘度的影響,一般說來,Zeta 電勢越大,雙電層越薄,粘度越小,電滲流值越大。

毛細管電泳
電滲在電泳分離中扮演著重要角色,是伴隨電泳產生的一種電動現象。多數情況下,電滲流速度是電泳速度的5-7 倍。因此,在毛細管電泳(CE)中利用電滲流可將正、負離子和中性分子一起朝一個方向產生差速遷移,在一次CE 操作中同時完成正、負離子的分離測定。由於電滲流的大小和方向可以影響CE 分離的效率、選擇性和分離度,所以成為優化分離條件的重要參數。電滲流的細小變化將嚴重影響CE 分離的重現性(遷移時間和峰面積)。所以,電滲流的控制是CE 中的一項重要任務。用來控制電滲流的方法主要有改變緩衝溶液的成分和濃度;改變緩衝溶液的pH 值;加入添加劑;毛細管內壁改性-物理或化學方法塗層及動態去活;外加徑向電場;改變溫度等。
中性物質可以用作測定電滲的標記物。例如二甲基甲醯胺(DMF)、二甲基亞碸(DMSO)、β-萘酚、丙酮、甲醇和乙醇等,均可作為電滲標記物。
1. 分離模式
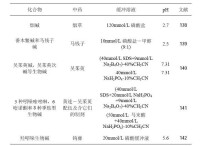
毛細管電泳根據分離模式不同可以歸結出多種不同類型的毛細管電泳,見表1。毛細管電泳的多種分離模式,給樣品分離提供了不同的選擇機會,這對複雜樣品的分離分析是非常重要的。
表1 毛細管電泳類型
類型縮寫說明1 單根毛細管毛細管區帶電泳CZE毛細管和電極槽灌有相同的緩衝液毛細管等速電泳CITP使用兩種不同的CZE 緩衝液毛細管等電聚焦CIEF管內裝pH 梯度介質,相當於pH 梯度CZE膠束電動毛細管色譜MECC在CZE 緩衝液中加入一種或多種膠束微乳液毛細管電動色譜MEEKC在CZE 緩衝液加入水包油乳液高分子離子交換毛細管電動色譜PICEC在CZE 緩衝液中加入可微觀分相的高分子離子開管毛細管電色譜OTCEC使用固定相塗層毛細管,分正、反相於離子交換親和毛細管電泳ACE在CZE 緩衝液或管內加入親和作用試劑非膠毛細管電泳NGCE在CZE 緩衝液中加入高分子構成篩分網路2 單根填充管毛細管凝膠電泳CGE管內填充凝膠介質,用CZE 緩衝液聚丙烯醯胺毛細管凝膠電泳PA-CGE管內填充聚丙烯醯胺凝膠瓊脂糖毛細管凝膠電泳Agar-CGE管內填充瓊脂糖凝膠填充毛細管電色譜PCCEC毛細管內填充色譜填料,分正、反相於離子交換等3陣列毛細管電泳CAE利用一根以上的毛細管進行CE 操作4晶元式毛細管電泳CCE 利用刻制在載玻片上的毛細通道進行電泳5 聯用毛細管電泳/質譜 CE/MS常用電噴霧介面,需揮發性緩衝液毛細管電泳/核磁共振CE/NMR需採用停頓式掃描樣品峰的測定方法毛細管電泳/激光誘導熒光CE/LIF具單細胞、單分子分析潛力
2. 操作方式

毛細管電泳
3. 分離通道形狀
按分離通道形狀分為圓形、扁形、方形毛細管電泳等。
4. 緩衝液的介質
根據配製緩衝液的介質的不同,可以把CE分為水相毛細管電泳和非水毛細管電泳(NACE)。NACE是以有機溶劑作介質的電泳緩衝液代替以水為介質的緩衝溶液,增加了疏水性物質的溶解度,特別適用於在水溶液中難溶而不能用CE分離的物質或在水溶液中性質相似難以分離的同系物,拓寬了CE的分析領域。
毛細管電泳通常使用內徑為25-100 μm 的彈性(聚醯亞胺)塗層熔融石英管。標準毛細管的外徑為375 μm,有些管的外徑為160 μm。毛細管的特點是:容積小(一根100 cm×75 μm 管子的容積僅4.4 μL);側面/截面積比大,因而散熱快、可承受高電場(100-1000 V/cm);可使用自由溶液、凝膠等為支持介質;在溶液介質下能產生平面形狀的電滲流。
毛細管電泳
(1)高效 塔板數目在105-106 片/m 間,當採用CGE 時,塔板數目可達107 片/m 以上;
(2)快速 一般在十幾分鐘內完成分離;
(3)微量 進樣所需的樣品體積為nL 級;
(4)多模式 可根據需要選用不同的分離模式且僅需一台儀器;
(5)經濟 實驗消耗不過幾毫升緩衝溶液,維持費用很低;
(6)自動 CE 是目前自動化程度較高的分離方法。
毛細管電泳的缺點是:
(1)由於進樣量少,因而製備能力差;
(2)由於毛細管直徑小,使光路太短,用一些檢測方法(如紫外吸收光譜法)時,靈敏度較低;
(3)電滲會因樣品組成而變化,進而影響分離重現性。
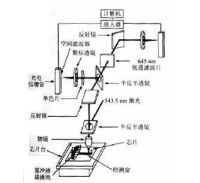
毛細管電泳系統的基本結構包括進樣系統、兩個緩衝液槽、高壓電源、檢測器、控制系統和數據處理系統,如圖1所示。
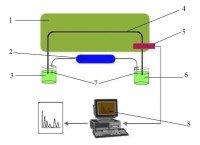
毛細管電泳
1-溫度控制系統;2-高壓電源;3-高壓電極槽;4-毛細管;5-檢測器;6-低壓電極槽;7-鉑絲電極;8-記錄/數據處理
檢測器
由於毛細管內徑的限制,檢測信號是CE系統最突出的問題。紫外可見法(UV)是CE常用的檢測方法,但是受到儀器、單波長等因素的限制。目前應用最廣泛的是二極體陣列(PDA)檢測器。常規的檢測器還有靈敏度很高的激光光熱(LIP)和熒光(FL)檢測器。近些年,在實際應用中還產生了激光誘導熒光(LIF)、有良好選擇性的安培(EC)、通用性很好的電導(CD)助以及可以獲得結構信息的質譜(MS)等多種檢測器。迄今為止,除了電感耦合等離子體(ICP)和紅外(IR)技術沒有和CE聯用,其他的檢測方法均和CE聯用並且大部分實現商品化。使用CE時應該根據所分析物質的特點,選擇相應分離模式和檢測器,以揚長避短,得到最佳分析效果。
1 緩衝液

毛細管電泳
2 pH值
緩衝體系pH的選擇依樣品的性質和分離效率而定,是決定分離成敗的一大關鍵。不同樣品需要不同的pH分離條件,控制緩衝體系的pH值,一般只能改變電滲流的大小。pH能影響樣品的解離能力,樣品在極性強的介質中離解度增大,電泳速度也隨之增大,從而影響分離選擇性和分離靈敏度。pH還會影響毛細管內壁硅醇基的質子化程度和溶質的化學穩定性,pH在4-10之間,硅醇基的解離度隨pH的升高而升高,電滲流也隨之升高。因此,pH為分離條件優化時不可忽視的因素。

毛細管電泳
在CE中,分離電壓也是控制電滲的一個重要參數。高電壓是實現CE快速、高效的前提,電壓升高,樣品的遷移加大,分析時間縮短,但毛細管中焦耳熱增大,基線穩定性降低,靈敏度降低;分離電壓越低,分離效果越好,分析時間延長,峰形變寬,導致分離效率降低。因此,相對較高的分離電壓會提高分離度和縮短分析時間,但電壓過高又會使譜帶變寬而降低分離效率。電解質濃度相同時,非水介質中的電流值和焦耳熱均比水相介質中小得多,因而在非水介質中允許使用更高的分離電壓。
4 溫度
溫度影響分離重現性和分離效率,控制溫度可以調控電滲流的大小。溫度升高,緩衝液粘度降低,管壁硅輕基解離能力增強,電滲速度變大,分析時間減短,分析效率提高。但溫度過高,會引起毛細管柱內徑向溫差增大,焦耳熱效應增強,柱效降低,分離效率也會降低。
5 添加劑
在電解質溶液中加入添加劑,例如中性鹽、兩性離子、表面活性劑以及有機溶劑等,會引起電滲流的顯著變化。表面活性劑常用作電滲流的改性劑,通過改變濃度來控制電滲流的大小和方向,但當表面活性劑的濃度高於臨界膠束濃度時,將形成膠束。加入有機溶劑會降低離子強度,Zeta電勢增大,溶液粘度降低,改變管壁內表面電荷分佈,使電滲流降低。在電泳分析中,緩衝液一般用水配製,但用水一有機混合溶劑常常能有效改善分離度或分離選擇性。
毛細管電泳
CE的常規進樣方式有兩種:流體力學和電遷移進樣。電遷移進樣是在電場作用下,依靠樣品離子的電遷移和(或)電滲流將樣品注入,故會產生電歧視現象,會降低分析的準確性和可靠性,但此法尤其適用於粘度大的緩衝液和CGE情況。流體力學進樣是普適方法,可以通過虹吸、在進樣端加壓或檢測器端抽空等方法來實現,但選擇性差,樣品及其背景同時被引入毛細管,對後續分離可能產生影響。通過進樣時間也可以來改善分離效果,進樣時間過短,峰面積太小,分析誤差大。進樣時間過大,樣品超載,進樣區帶擴散,會引起峰之間的重疊,與提高分離電壓一樣,分離效果變差。
另外,毛細管電泳技術的高分離性能以及消耗試劑少等特點使其分析領域得到了廣泛的應用,但是其常規分析的靈敏度不能適應痕量分析的要求,限制了它的應用和推廣。樣品前處理技術可以提高樣品通量或將痕量分析物進行預富集,去除樣品基質,將其與毛細管電泳技術聯用不僅可以提高分析的靈敏度,同時也消除了大部分可能的基質干擾,是一種比較理想的富集分離檢測技術。常用的有CE一流動注射聯用技術、固相萃取-CE聯用技術、固相微萃取-CE聯用技術、液相微萃取-CE聯用技術、微透析-CE聯用技術和膜萃取-CE聯用技術。
生物體內,蛋白質是必不可少的生命物質,是藥物的重要靶點之一。研究藥物與蛋白質之間的相互作用,有助於了解藥物在體內的運輸和分佈的情況,對於闡明藥物的作用機制、葯代動力學以及藥物的毒性都有非常重要的意義。藥物分子與蛋白質分子相互結合的主要部位是蛋白質上的鹼性氨基酸殘基,相互作用力主要有靜電作用、氫鍵、疏水作用、范德華力和電荷轉移作用,通常藥物與蛋白之間的相互作用並不是一種作用力的單獨作用,而是多種作用力的協同作用。這種相互作用的量化參數就是藥物與蛋白的結合常數Ka。
結合常數Ka 的測定方法現主要有:熒光光譜法,紅外光譜法,毛細管電泳法,核磁共振法,以及電化學等方法。其中毛細管電泳法以其效率高,速度快等優點,已被較多採用。Scatchard 模型是現在公認的測定藥物與蛋白結合參數的理論模型。
區帶毛細管電泳法是利用蛋白質與藥物的結合產物同遊離的藥物或蛋白的淌度差異來研究相互作用的。
毛細管電泳
Olivares,Smith和Henion等分別在1987-1988年提出毛細管電泳-質譜聯用(CE-MS)技術,在CE中,紫外檢測器由於通過樣品的光程較短導致靈敏度較低,特別對一些紫外吸收較弱的化合物的檢測。近年由於大氣壓電離(API)、電噴霧電離(ESI)及新型質譜儀的快速掃描等新技術的出現,足以滿足CE窄峰形的特點,使得CE-MS,CE-MS-MS均得到快速發展,並正在成為實驗室的重要常規分析方法之一。
質譜檢測器有如下特點:

毛細管電泳
2)由於質譜的選擇性和專一性,彌補了樣品遷移時間變化的不足;
3)質譜檢測的靈敏度優於紫外分光光度法;
4)質譜在檢出峰的同時還能給出分子量和結構信息;
5)某些質譜技術可以給出多電荷離子,對分析大分子如糖,蛋白質等與CE聯用更有利。
CE的許多模式,如CZE,MEKC,CITP,CGE和ACE以及CEC等都能與質譜檢測器成功地連接,其中應用較多的仍是CZE-MS。MEKC由於添加表面活性劑形成的膠束會抑制樣品離子的信號,所以MEKC-MS使用較少。與CE相連的MS最常用的電離方式是ESI,可以直接把樣品分子從液相轉移到氣相,而且可以測定分子量較大的樣品。與CE相連的質譜儀主要有三元四極(QQQ)質譜儀,離子阱(TTT)質譜儀,傅立葉轉換離子回旋加速器共振(FT-ICQ)質譜儀和飛行時間(TOF)質譜儀等,前兩者較為常用。CE-MS常用的介面有無套管介面,液體接合介面和同軸套管流體介面等三種,后兩種介面均在毛細管流出部分引入補充流體,以維持一個穩定的電噴霧流。CE-MS所使用的緩衝液最好是易揮發、低濃度,可獲得較好的離子流響應。與質譜相連的CE中常使用加入較高含量的有機溶劑(例如甲醇、乙睛)的緩衝液或者使用非水毛細管電泳,有利於離子噴霧過程,可以增進檢測的靈敏度。天然植物葯各組分之間以及藥物與其代謝產物之間往往結構比較相似,用CE-MS無論對分離及鑒定都顯示了優勢。Henion等用同軸套管流體介面,全掃描質譜方式分離了8種合成的異哇琳生物鹼,對威氏黃柏(Phenllodendron wilsonii)樹皮中的8種組分進行了分離並鑒定了其中的6種。Unge等用同樣介面將盧竹鹼等巧種叫垛類生物鹼和生物胺基線分離。此外,離子噴霧(Ionspray,ISP),大氣壓化學電離(APCI)等也被應用在CE-MS中。
1990年瑞士Ciba-Geigy公司的Manz和Widmer首次提出微全分析系統

毛細管電泳
微流控分析系統可大大提高分析速度和極大地降低分析費用,微流控CE分析系統通常也被稱為集成毛細管電泳(Integrated capillaryelectrophoresis,ICE)。微流控分析系統開創了分析科學歷史的新篇章,使分析科學進入了一個微型化、集成化和自動化的嶄新世界。
CE具有多種分離模式(多種分離介質和原理),故具有多種功能,因此其應用十分廣泛,通常能配成溶液或懸浮溶液的樣品(除揮發性和不溶物外)均能用CE進行分離和分析,小到無機離子,大到生物大分子和超分子,甚至整個細胞都可進行分離檢測。它廣泛應用於生命科學、醫藥科學、臨床醫學、分子生物學、法庭與偵破鑒定、化學、環境、海關、農學、生產過程監控、產品質檢以及單細胞和單分子分析等領域。
毛細管電泳
1 CE在藥物製劑分析中的應用
藥物製劑中成分複雜,除含有有效成分外,往往還含有一些有效成分的穩定劑或保護劑,一般幾毫克的有效成分需要幾十毫克的基體。CE法具有能排除高含量複雜基體干擾、檢測痕量成分的能力,且樣品只需經簡單預處理即可分析其有效成分含量,現已廣泛應用於片劑、注射劑、糖漿、滴耳液、乳膏劑及復方製劑等各種劑型中主葯成分的定量測定。
2 CE在藥物雜質檢查中的應用
藥物合成中帶入的雜質和藥物的降解產物通常與藥物有相似的結構,而且一般含量很低。CE作為藥物的雜質痕量組分分析方法,具有多組分、低含量和同時分離分析能力,故可以用毛細管電泳作為藥物雜質的檢測手段。CE也可以用於藥物生產過程全方位控制與檢測,以保證藥物質量,提高工藝水平。己有文獻報道用NACE法測定己烯雌酚片及其降解物;CE法定量分析鹽酸羅匹尼羅及其潛在雜質;CZE法分析伊班磷酸鹽及其相關雜質;CZE和CITP法檢測高舍瑞林中縮氨酸和反離子物質的含量;CZE法定量檢測半胱胺鈉磷酸鹽中的雜質。
毛細管電泳
中藥品種繁多、藥材產地各異、成分複雜,無論是藥材還是成藥的分析,都是一項非常艱難的任務。中藥分析工作用現代化儀器設備和科技手段(如薄層色譜、HPLC等)雖取得巨大進展和成就,但往往只是對藥材和成藥成百上千個成分中的一個或幾個成分的分析,實際只是一種象徵性的代表式分析,與之起化學和藥理效應的實際組合成分(起碼是有效成分)相比,仍有相當大的距離。隨著CE技術對中藥材及其有效成分的鑒別與分析的快速發展,建立在此基礎上的中成藥和中藥復方製劑中有效成分的定性、定量分析已有進展,且有希望解決長期困擾中藥質量控制中的重大難題。近年,報道CE分析中藥材已有18種、成藥70種和有效成分120個以上。
4 CE在手性藥物分析中的應用
毛細管電泳
5 生物樣本中的藥物及其代謝產物分析
生物體內藥物及其代謝物的隨時間與位置分佈研究,即藥物動力學分析,在臨床醫學中有重要意義。在非水溶劑中可降低被分析物與管壁的作用,降低由於吸附所引起的峰拓寬並改善拖尾,同時可顯著提高被分析物的回收率,降低用管壁面積較大的毛細管進行分析時被分析物的損失。近年來,用毛細管電泳法進行生物樣本中的藥物及其代謝產物的分析已成為研究熱點。已有文獻報導用CE法監測腺昔及其代謝物含量變化;用CE法測定人血漿中的優降糖、甲福明二甲雙肌、苯乙雙肌含量;用CE一化學發光法檢測人尿中兒茶酚胺的含量;用CZE-安培法測定尿中的L-酪氨酸及其代謝物濃度;用CZE法測定人尿中兩種巴比妥鹽的濃度;HPCE法測定頭抱克羅血漿濃度;CE法測定血漿中蛋氨酸的含量;用CE-電導法檢測血清中的丙戊酸含量。
CE分析速度快,有良好的時間分辨性,能為治療機制與用藥水平提供可靠的分析,將來一定會加深在此領域內的應用。

毛細管電泳
CE技術的研究和應用,給藥物分析領域和藥品檢驗工作帶來了生機與活力,無疑將對該專業技術的發展及提高起著重要的推動和促進作用。尤其以對基因工程藥物、中成藥復方製劑的分析和中藥材種屬的鑒定,令人矚目。但任何事物都有兩面性,它也有弱點和不足,如有的藥物用CE分析精確度還不夠高;有的靈敏度很高,但專屬性界定尺度又不易掌握;CE儀器昂貴,很難普遍推廣等等,都需要不斷研究解決。尤其以如何巧妙地與其它方法和技術(如HPLC、MS等)聯合使用,以收到更好的效果,是今後CE技術研究、完善的方向和課題。可喜的是,這方面的工作已開始啟動,CE一HPLC、CE一MS聯用己取得高效率、高質量的分析成果。經過科學工作者的不懈努力,一個藥物分析領域的新技術快速發展時期即將到來。
[1] 朱建中,周衍. 電化學生物感測器的進展[J]感測器世界, 1997,(04).
[2] 呂亞萍,周永列. 高效毛細管區帶電泳法快速測定尿液中的肌酐[J]分析試驗室, 2006,(02).
[3] 蔡梅,安登魁. 毛細管電泳的柱上濃縮技術[J]國外醫學。藥學分冊, 1999,(01).
[4] 陳華,章竹君,付志鋒. 流動注射化學發光法測定撲熱息痛[J]分析化學, 2002,(11).
[5] 陳建,朱化雨,陳常興. 分光光度法測定血清中的葡萄糖[J]分析儀器, 1998,(02).
[6] 王錚,張惠新. 聚碸中空纖維超濾膜的製備及應用[J]工程塑料應用, 2002,(08).
[7] 周偉紅,吳明嘉,汪爾康. 高效毛細管電泳安培檢測的進展[J]分析化學, 1995,(03).
[8] 吳明嘉,許莉娟. 毛細管電泳安培檢測裝置的研究[J]分析化學, 1995,(05).
[9] 李慧琴. 微透析技術及其在生化分析中的應用[J]解放軍預防醫學雜誌, 2000,(03).
[10] 趙燕燕,楊更亮,李海鷹,陳義. 高效前沿分析的發展及在藥物-蛋白結合研究中的應用[J]化學通報, 2003,(05).
[11] 袁道強. 毛細管電泳法測定奶粉中的鎘、鉛和銅[J]食品科學, 2002,(01).
[12] 李玉珍,尹洧. 毛細管電泳在生命科學研究中的應用(1)[J]生命科學儀器, 2005,(03).
[13] 楊冰儀,莫金垣. 肉類中己烯雌酚的高速毛細管電泳安培法測定[J]分析測試學報, 2003,(03).
[14] 舒友琴,袁道強. 飲料中六種金屬陽離子的毛細管電泳分析[J]食品工業科技, 2000,(02).
[15] 張國華,王延琮,張永友,王磊,張玉奎. 高效毛細管電泳測定黃連及成藥中小檗鹼型生物鹼的含量[J]色譜, 1995,(04).
[16] 丁永生,林炳承. 人血清白蛋白與手性藥物相巨作用的毛細管電泳研究Ⅰ.液相預柱毛細管電泳技術定量可靠性的考察[J]色譜, 1999,(02).
[17] 劉志松,方肇倫. 高效毛細管電泳在藥物分析中的應用[J]色譜, 1996,(05).
[18] 周大煒,李樂道,李發美. 藥物-蛋白結合作用的分析方法研究[J]色譜, 2004,(02).
[19] 潘文. 毛細管電泳電化學發光在藥物檢測方面的應用研究[D]湖南大學, 2007.
[20] 郭岳. 黃連及復方中藥生物鹼有效性的毛細管電泳分析[D]吉林農業大學, 2006.
[21] 李美林. 稀土離子與微過氧化物酶-11相互作用機理的研究[D]南京師範大學, 2006.
[22] 周維. 毛細管電泳技術在生物醫藥方面的應用研究[D]汕頭大學, 2005.
[23] Chester T.L,Pinkston J.D, Raynie D.E. Super critical fluid chromatography and extraction .Anal. Chem, 1998, 70 (12) :301-319 .
[24] Gaillard Y,Pepin G. Gas chromatographic-mass spectrometric quantitation of dextro- propoxyphene and norpropoxyphene in hair and whole blood after automated on-line solid phase extraction. Application in twelve fatalities .J. Chromatogr. B, 1998, 709 (1) :69-77 .
[25] Debets A.J.J,Mazereeuw M., Voogt W.H. et al. Electrophoretic sample pre-treatment techniques coupled on-line with column liquid chromatography .J. Chromatogr. A. 1992, 608 :151-158 .
[26] Quirino J.P,Terabe S. On-line concentration of neutral analytes for micellar electrokinetic chromatography I. Normal stacking mode .J. Chromatogr. A. 1997, 781 :119-128 .