SMA
脊髓性肌萎縮症
但需要注意的是每個SMA患者的病程可能都會有所差異。 Ⅲ型SMA患者手指伸出後會有細微的顫動,但舌部肌肉震顫徠的情況較罕見。有時SMA患者的SMN1基因不存在缺失,但卻存在突變的情況。
脊髓性肌萎縮症(SMA),是一類由脊髓前角運動神經元變性導致肌無力、肌萎縮的疾病。屬常染色體隱性遺傳病,臨床並不少見。本病臨床表現差異較大,根據患者起病年齡和臨床病程,將SMA由重到輕分為4型。共同特點是脊髓前角細胞變性,臨床表現為進行性、對稱性,肢體近端為主的廣泛性弛緩性麻痹與肌萎縮,智力發育及感覺均正常。
脊髓性肌萎縮症(spinal Muscular atrophy 縮寫:SMA)是一種運動神經元性疾病。運動神經元對用於進行爬、走、頭頸控制以及吞咽等這一類活動的隨意肌會產生影響。SMA是一種相對常見的“罕見病”——新生兒患病率約為1:6000,每40名成人中就有1人是致病基因攜帶者。
儘管SMA對於近端肌(最靠近人體軀幹部的肌肉,即肩、髖和背部)所造成的影響最為嚴重,但其實它對人體全身的肌肉都可以造成侵害。通常患者腿部無力的情況較手臂處更為嚴重,進食和吞咽有時也會受到影響。呼吸肌(參與呼吸與咳嗽的肌肉)的受累會導致患者更容易出現肺炎以及其它的肺部問題。SMA患者的感官系統不會受到影響,智力正常,而且他們通常都較為聰明、隨和。基於一些特定的關鍵性運動功能指標,患者通常會被劃分為4個類型。
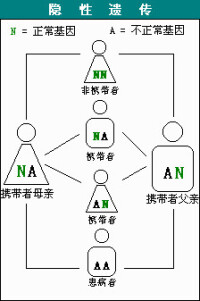
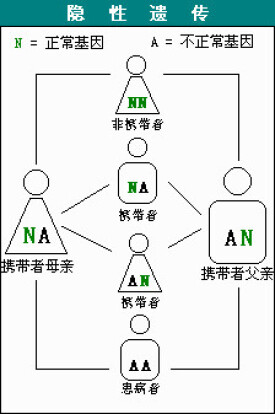
SMA是一種常染色體隱性遺傳疾病。父母雙方必須都是致病基因攜帶者,並且都把這一基因傳給孩子,孩子才有可能染病——而且每個孩子染病的幾率是25%。
人體內的一種基因(SMN1 —— “運動神經元生存1號”基因)可以製造出被稱為“運動神經元生存”(SMN)的蛋白。SMA患者體內的這種基因要麼缺失要麼就是存在變異,而由此所導致的SMN蛋白缺乏會對運動神經元產生最為嚴重的影響。運動神經元是位於脊髓內的神經細胞,由它們所延伸出來的神經纖維通向身體各處的肌肉。SMN對於運動神經元的生存和健康極其重要,沒有這種蛋白,神經細胞就有可能萎縮並最終死亡,從而導致肌肉的無力。
隨著SMA患兒的生長,他們的身體要承受雙重的壓力,首先是運動神經元的減少,其次是神經和肌肉細胞需求的增長。由此而導致的肌肉萎縮會造成肌肉無力以及骨頭和脊柱的變形,這些有可能又會造成進一步的功能喪失,並危及他們的呼吸系統。
按照患者所能達到的身體功能指標,SMA被分為Ⅰ、Ⅱ、Ⅲ、Ⅳ4個類型。但需要注意的是每個SMA患者的病程可能都會有所差異。
徠
Ⅰ型SMA也被稱作沃德尼格·霍夫曼病(Werdnig-Hoffmann Disease)。患兒通常在6個月內可以被確診,而大多數病例是還不滿3個月即被檢出。一些母親在孕期的最後幾個月中甚至就會察覺到有胎動減少的情況。
通常Ⅰ型患兒根本無法抬頭或是達到嬰兒早期所應該具有的正常運動功能。他們的頭部控制能力很差,無法像正常孩子那樣有力地踢腿或站立。沒有支撐,他們就無法坐穩。吞咽和餵食可能會有困難,通常到一定的時候都會出現這樣的情況。孩子無法很好地控制自己口腔內的分泌物。舌頭會有萎縮、波動或細微顫動的情況,這也稱之為肌束震顫。肋間肌可以幫助擴張胸腔,但這些肌肉的無力使得孩子的胸腔體積比正常情況要小。SMA患者最有力的呼吸肌是橫膈膜,這就使得他們好像是在用腹部肌肉呼吸一樣,這種隔式(腹式)呼吸會使患者胸部出現凹陷的癥狀。同樣這種呼吸方式也會導致他們的肺無法完全發育,咳嗽會非常微弱,睡眠中也難以進行有足夠深度的呼吸來維持體內正常的氧含量。
Ⅱ型
Ⅱ型SMA患兒幾乎在2歲前都可以被確診,而大多數病例在15個月內即可被檢出。儘管此型患兒通常無法自己進入坐姿,但若幫助其獲得坐姿后,他們也可以獨立坐穩。某些情況下,在外力、支具或站立架的輔助下他們還可以站立。Ⅱ型SMA患兒吞咽方面的問題通常並不突出,但孩子之間卻會存在個體差異。有些患者可能無法自行攝入足夠的食物以維持正常的體重和生長所需,這時可能就需要使用鼻飼管來餵養。Ⅱ型SMA患兒經常會有舌部肌肉震顫以及手指伸出后出現細微顫動的情況。他們肋間肌比較虛弱,所以使用的是隔式呼吸。他們咳嗽困難,而且睡眠時可能由於呼吸深度不夠而難以維持體內正常的氧含量。隨著孩子的生長,他們幾乎都會出現脊柱側彎的情況,這就使得在必要的時候要進行脊柱手術或使用相應的支具。骨密度降低會使得他們更容易骨折。
Ⅲ型
Ⅲ型SMA通常也被稱作庫傑爾博格·偉蘭德病(Kugelberg-Welander)或少年型脊肌萎縮症。它的發病年齡更為寬泛,雖然3歲前獲得診斷較為常見,但早至1歲,晚至青春期都可能發病。Ⅲ型SMA患者可以獨自站立并行走,但隨著病程的發展,某個時候行走就會出現困難。早期所應該達到的運動指標都正常,然而,一旦開始行走他們就會經常摔倒,俯身或坐在地上難以站起,可能也無法奔跑。Ⅲ型SMA患者手指伸出後會有細微的顫動,但舌部肌肉震顫的情況較罕見。餵食和吞咽的問題在他們童年期間並不常見。有些患者可能會在童年、青年甚至成年時喪失行走能力,但這經常都和他們身體的快速發育或其它疾病有關。
Ⅲ型 成年發病
成年型患者的癥狀通常始於35歲后。SMA很少於18-30歲之間發病。成年型SMA較其它類型要少見得多,被定義為要18歲后才出現無力的癥狀,但大多數報道的Ⅳ型病例都是在35歲后發病。其典型特徵是隱形發病,病情發展非常緩慢。延髓所控制的肌肉,即用於吞咽和呼吸的肌肉很少受到侵害。
SMA患者的身體功能通常會隨著時間的推移而喪失,這在他們身體快速發育或患病期間顯得較為突出,但基於目前的研究這種現象還無法解釋。儘管隨著年齡增長患者身體功能的喪失是普遍的趨勢,但據觀察他們在較長的時間段內(常會有數年之久)身體功能會非常穩定。
SMA的診斷主要通過驗血檢測SMN1基因是否存在,並配合體檢及了解家族病史來進行。
正常情況下,人體內擁有兩個分別被稱為SMN1和SMN2的基因。約95%的SMA患者存在SMN基因序列的缺失,而健全人則不存在這種情況。有時SMA患者的SMN1基因不存在缺失,但卻存在突變的情況。SMN2是SMN1的備份基因,兩者幾乎完全相同,SMN2的拷貝數量關係到SMA疾病的嚴重程度,但並不能據此來推測患者屬於第幾型。SMA的分型通常需要經過臨床檢查,評估孩子肌肉無力的程度以及所能達到的主要運動功能指標(比如獨自坐穩或行走)后才能確定。
偶爾,醫生也會要求進行肌肉活檢或肌電圖檢查。自從血液基因測試開始應用之後,肌肉活檢幾乎就不再使用了,除非血液DNA測試結果為陰性。
肌電圖能夠測量肌肉電活動的情況。有時,這項檢測會被用來鑒別與SMA類似的其它神經或肌肉疾病。細小的電極(針)插入患者手臂和大腿處的肌肉后,儀器上就可以顯示相應的電子圖形並將其記錄下來。此外,進行神經傳導速度測試(NCV)還可以幫助評估神經在電刺激下的反應情況。醫生對患者不斷施以非常輕微的電擊以幫助評估神經的功能和完整性。如果孩子需要接受這項測試,可能的話應盡量由具備相關看護經驗的醫生來進行。
研究人員已經確定,SMN蛋白主要依靠SMN1基因來製造。而這個SMN1基因的缺失或缺陷正是導致SMA的原因所在。不過,人體內還有另外一個與SMN1類似的被稱為SMN2的基因,只是它無法像SMN1那樣製造足夠多或正常的蛋白。確定預后的一種方法就是看患者體內SMN2基因的拷貝數量有多少。數量越多的話,其製造的SMN蛋白也就會越多,更多的運動神經元才更有可能保持健康。只有1個或2個SMN2基因拷貝的患者通常SMA的癥狀表現得最為嚴重,而擁有3個或更多拷貝數量的患者癥狀通常都會較輕一些。
每一型SMA患者之間都會存在個體差異,所以當需要考慮個人護理問題的時候,這一點應當加以注意。
撫養SMA患兒與撫養其他孩子不應該有什麼不同。應儘可能做符合其年齡段的事情。往往這會意味著要進行一些讓孩子能夠適應的調整。但非常重要的一點就是要幫助他們達到自身最大的潛能。
您需要明白的很重要的一點就是家長和患者都有自己的權利,你們並不是孤立無援的。大多數的醫院都設有社會服務部,你們可以向其尋求幫助。如果覺得事情不對勁,不要害怕說“不”。也不要有什麼擔心而不敢提出問題。如果忘記詢問什麼事情,可以打電話給你的醫生或聯繫FSMA尋求建議。基於這一點,有熟悉SMA及其併發症的醫生追蹤關注您的孩子是非常重要的。
Ⅰ型及某些Ⅱ型
大部分Ⅰ型SMA患兒獲得診斷時都還是嬰兒,所以在協助他們身體、情感及認知的發展上有很多事情是我們可以做的。用氣球和羽毛當作玩具可以很好地刺激他們,讓他們獲得一種獨立感和成就感。伸手夠物的遊戲對孩子很有幫助,常在物理或職業療法中被使用。不論孩子多小,來自持證物理或職業治療師的相關指導和建議都是很重要的。他們還可以推薦適合的座位系統,在保證舒適和最佳運動性的前提下為孩子提供最大的幫助。
由於水的浮力可以讓孩子進行在岸上無法完成的手和腿部活動,所以水療對他們來說是很有幫助的。不過要確保水溫至少有32℃,而且孩子的頭部始終在水面上。您必須時刻看護,避免孩子嗆水(將水吸入肺中)。
吞咽有困難的SMA患兒吃東西時會有將異物吸入肺部的危險。有時他們還可能將自己口腔內的分泌物吸入肺中。孩子進食時可能會被嗆到,而且隨著吞咽困難的加重,他們的體重也可能會下降。這時也許就需要輔助餵食了,下面是兩個可供選擇的方法:
1. 鼻胃管(NG-Tube):一根通過鼻子直接插入胃中的管子
2. 胃造口飼餵管(G-Tube):一根通過外科手術穿過腹部皮膚直接插入胃中的管子
因為Ⅰ型SMA患兒咳嗽困難,所以聯繫一位呼吸治療師是非常重要的,他能教您如何給孩子進行胸腔理療(CPT)。CPT是一種通過調整身體姿勢,拍打胸廓幫助鬆動肺內分泌物來清理聚集在肺部黏液的方法。唾液停留在鼻咽處就會造成輕微的咕嚕聲。通常這些黏液或分泌物需要藉助吸痰設備來清除。吹嘴或吹泡泡可以促進改善孩子呼吸肌的力量。
使用輔助咳痰設備對SMA患者也會有所幫助。CoughAssist(TM)咳痰機可以使用正向壓力經氣道由外至內將空氣送入患者肺內使肺部膨脹起來,然後快速轉為由內至外的反向壓力,將肺內的空氣抽出來。這種快速的壓力轉變可以產生很大的呼氣氣流,從而模擬出咳嗽的效果。這種被稱為“機械式充氣-排氣”的技術避免了清理肺內分泌物時對氣道的傷害。此設備無需藉助侵入式器械和方法就可以提高患者的舒適度和生活質量。成功使用CoughAssist(TM)咳痰機的患兒年齡最小的只有4個月大。
使用脈搏血氧儀測量血液中的氧飽和度就可以對患者是否存在呼吸困難進行監測。將一個帶有紅燈和感測器的夾子或膠帶固定在患者的手指或腳趾上就可以測量氧飽和度了。Ⅰ型SMA患兒睡覺時往往都需要給予呼吸支持。有些孩子,尤其是在感冒期間,會需要更多的呼吸支持。以下是幾種可以考慮的選擇:
1. BiPAP(雙水平氣道正壓通氣)藉助一個像帽子的裝置將通氣鼻罩固定在患者的鼻子上。患者吸氣時,BiPAP可以將更多的空氣送入他們的肺中,使其肺部充盈的程度較自主呼吸時更高。而呼氣時,BiPAP會降低壓力,患者肺內的空氣就會被動地排出。BiPAP呼吸機可以同步偵測到患者何時吸氣和呼氣,還可以設定呼吸頻率以保證每分鐘為患者提供一個最低次數的呼吸。患者呼吸的次數可以高於所設定的頻率,BiPAP將會適時地提供更多次數的呼吸。SMA患者一定不能使用CPAP(持續氣道正壓通氣)。
2. 負壓通氣指的是用類似早期“鐵肺”那樣的一個大型倉體或罐體環繞患者胸腔以提供呼吸支持的方式。這一倉體與一台真空泵相連,倉內的空氣被抽走之後,患者的胸廓就會擴張,隨之將空氣帶入肺中。Port-A-Lung就屬於一種負壓通氣機,它可以設定真空壓力的大小以及每分鐘支持患者呼吸的次數。
3. 機械通氣機或呼吸器有很多不同的類型。機械通氣機更為複雜,但也可以對更多的可變因素加以控制,呼吸的頻率和深度都可以進行設定。患者清醒時,機械通氣可以通過鼻罩或口含器進行,否則就需要通過氣管造口插管來實現。氣管造口是用手術的方式在頸部氣管處開一個口以便插入呼吸導管。氣管造口導管不經過口腔和聲帶而是穿過頸部皮膚直接進入到氣管內。通氣機或呼吸器都是連接在口鼻氣管導管或氣管造口導管上的。
具體事宜,請向您的醫生或呼吸治療師進行諮詢。
當需要做出維持孩子生命的決定時,明白你們自身的權利是很重要的。針對這一極為敏感的話題,父母雙方一定要將自己的感受表達出來。這不是一個輕易可以做出的決定,所有的選擇都應當考慮到。同您醫院社會服務部的諮詢顧問進行交通也許會有所幫助。一旦做出了決定,請務必將其書面化,以便有關的醫務人員和家庭成員能夠清楚你們的意願。這是你經過慎重的考慮和痛苦的掙扎后才做出的決定,所以無論在什麼情況下,都不要讓別人來評斷你或是將他們的價值觀強加於你。
Ⅱ型及某些Ⅲ型
儘早讓孩子直立起來非常重要。站立對於他們的發育很重要。這可以讓他們獲得更好的呼吸和腸胃功能,也可以促進他們的運動能力。為了讓孩子能夠直立起來,父母首先要持支持的態度,有時還需要鼓勵醫生開出處方以便能獲得輔助孩子站立的設備。
挑選適合的輔助站立設備有幾個選擇可以考慮。
首先就是站立架和/或站立行走架。如果需要更多的活動性和獨立性的話,可站立式輪椅會比較理想,最小13個月大的孩子都可以使用。另外,支具也是一種選擇。重心移動式步行矯形器(RGO's)和承重式膝踝足矯形器(KAFO's)被發現對於Ⅱ型患兒都會有所幫助,那些使用的孩子都能夠走上幾步。使用合適的輔助器械或帶支具的行走設備是非常重要的,在治療師的指導下應當嘗試各種不同的選擇。
使用輕便的手動輪椅可能會令SMA患兒興奮不已。這讓他們藉助自身的力量就能獲得獨立,進行移動並嘗試“冒險之旅”。然而家長需要明白的一點是,孩子要獲得真正的獨立和移動性還是需要使用電動輪椅。
到了一定的時候,基本上所有Ⅰ型和Ⅱ型以及一些Ⅲ型的患兒都會出現脊柱側彎的情況。該如何應對主要要視側彎的嚴重程度而定。由於脊柱側彎會限制患者的呼吸和肺部功能,所以應當儘早採取防範措施。應對的選擇包括:量身定做座位系統、添加座位輔助裝置和配帶矯形背心。再往後可能就需要考慮進行脊柱融合手術了。
如果您的孩子經常感冒,咳嗽困難,您可能會想要了解輔助咳嗽設備的有關情況。在前面Ⅰ型患者的部分,您可以看到CoughAssist(TM)咳痰機的詳細介紹。
對於任何一個處在生長發育階段的孩子來說,飲食都是非常重要的。您孩子的飲食需要給予精心的照顧。身體超重會讓他們移動更加困難。就此與您的醫生和營養師保持溝通是非常重要的。
Ⅲ型
由於Ⅲ型患兒在一定的時間範圍內是能夠獨立行走的,所以對他們進行觀察是非常重要的,這樣可以儘早發現他們所遇到的困難。他們可能需要使用助行器或支具,稍遠的距離則可以考慮輕便的手動輪椅、電動車或其它的機動設備。這些應當向物理及職業治療師進行諮詢,此外他們的飲食也同樣需要加以注意。
Ⅳ型(成人型)
作為成人,您能夠明白自身的疾病及由此所帶來的限制。您應當與您的醫生,物理及職業治療師合作共同制定最適合您的治療計劃。同Ⅰ、Ⅱ、Ⅲ型患者一樣,飲食及營養也是您保持良好狀態的重要因素。
經過血檢確認是致病基因攜帶者之後,您可能已經諮詢過這方面的信息了。FSMA建議您向遺傳學顧問尋求建議。他們能幫助您更好地了解後代患病的可能性以及相關的風險。他們會了解您完整的家族史,包括每一名成員出現的疾病、死亡、死因、死胎以及流產的情況。如果您有孩子已經被診斷為SMA,那麼針對將來希望再次懷孕的事宜,遺傳學顧問會同您討論您需要考慮的選擇。
當前能夠獲得的信息顯示,SMA產前診斷的準確率是98%,是否進行這樣的檢測是非常個人化的決定,作出決定前父母雙方進行深入的溝通非常重要。
1.對稱性進行性近端肢體和軀幹肌無力肌萎縮,不累及面肌及眼外肌,無反射,亢進感覺缺失及智力障礙。
2.家族史符合常染色體隱性遺傳方式。
3.血清肌酸激酶(CK):患者血清CK水平正常或少數輕中度升高。
4.肌電圖顯示廣泛神經源性損害。
5.肌活檢顯示神經源性病理改變。
6.基因檢測:多重連接探針擴增法(MLPA)、實時熒光定量 PCR(qPCR)、PCR限制性酶切分析法(DHPLC)和變性高效液相色譜等檢測SMNl基因第7或第7、8外顯子純合缺失突變。MLPA、qPCR和DHPLC可用於SMNl和SMN2基因拷貝數檢測,酶切法可以用於sMNl基因外顯子7、8的純合缺失檢測;SMNl基因測序用於檢測SMNl基因內是否存在微小突變。
1.藥物治療
1)2019年2月,國家藥品監督管理局正式批准諾西那生鈉注射液用於治療5qSMA。5qSMA是該疾病最常見的形式,約佔所有SMA病例的95%。
2)5月24日,美國食品和藥物管理局(FDA)批准了諾華公司首款治療小兒脊髓性肌肉萎縮症的基因治療藥物Zolgensma上市。
3.預防或治療SMA的各種併發症
預防肺部感染及壓瘡、營養不良、骨骼畸形、行動障礙和精神社會性問題。
如伴有呼吸功能不全,需用人工呼吸器,保證氣道通暢,改善呼吸功能。長期卧床可造成墜積,誤吸也可造成肺炎。預防肺炎的有效措施有輔助咳嗽、胸部叩擊治療及間歇正壓通氣,即使在沒有急性呼吸道感染的情況下,患者也需保持良好的肺部通氣狀態,預防發生進行性肺不張。一旦有效肺活量下降,即使肢體或軀幹的肌力無明顯改變,發生肺炎的危險性也會增高。
4.康復治療
由於當前可治療SMA的藥物尚不能廣泛使用,因此,定期物理治療(PT)、正確使用支具或矯形器、規律運動訓練等積極的康復治療仍是目前干預、延緩疾病進展的主要手段。即使今後應用“可治療藥物”,康復訓練仍應貫穿治療全程。
1)60%~90%的1型和2型SMA患者在兒童早期出現脊柱側凸並持續發展,伴有不同程度的胸椎后凸,應常規作臨床脊柱檢查、正側位脊柱全長X片檢查。如脊柱側彎角度大於20度時應每6個月複查1次,直至骨骼發育成熟后每年複查1次。側彎角度大於20度時建議使用脊柱矯形器。是否採用手術干預主要取決於脊柱側彎程度 (主彎Cobb角≥50度)和進展速度(每年≥10度)。
3)呼吸功能訓練:包括呼吸肌肌力訓練、維持胸廓順應性訓練、咳嗽和排痰訓練等。
隨著病情的進展,肌無力可進一步導致骨骼系統、呼吸系統、消化系統及其他系統異常,其中呼吸衰竭是最常見的死亡原因。
有脊髓性肌萎縮症家族史的人群,生育前應進行產前診斷。
基本信息
- 類型
- 運動神經元性疾病
- 別稱
- 脊肌萎縮症
- 就診科室
- 神經內科
- 常見病因
- 常染色體隱性遺傳
- 常見癥狀
- 肌無力、肌萎縮