常染色體隱性遺傳病
由位於常染色體上的隱性致病基因引起的疾病
常染色體隱性遺傳病(autosomal recessive disorder)致病基因在常染色體上,基因性狀是隱性的,即只有純合子時才顯示病狀。此種遺傳病父母雙方均為致病基因攜帶者,故多見於近親婚配者的子女。
子代有1/4的概率患病,子女患病概率均等。許多遺傳代謝異常的疾病,屬常染色體隱性遺傳病。按照“一個基因、一個酶”(one gene one enzyme)或“一個順反子、一個多肽”(one cistron one polypeptide)的概念,這些遺傳代謝病的酶或蛋白分子的異常,來自各自編碼基因的異常。常見的常染色體隱性遺傳病有溶酶體貯積症,如糖原貯積症、脂質貯積症、粘多糖貯積症;合成酶的缺陷如血γ球蛋白缺乏症、白化病;苯丙酮尿症、肝豆狀核變性(Wilson病)及半乳糖血症等。
常染色體隱性遺傳病(autosoml recessive inheritabledisease)是由位於常染色體上的隱性致病基因引起的,其特點是:
①患者是致病基因的純合體,其父母不一定發病,但都是致病基因的攜帶者(雜合體)。
②患者的兄弟姐妹中,約有1/4的人患病,男女發病的機會均等。
③家族中不出現連續幾代遺傳,患者的雙親、遠祖及旁系親屬中一般無同樣的病人。
④近親結婚時,子代的發病率明顯升高。
通俗的來講,人體中每個細胞核中的常染色體有22對,每對染色體的DNA上有無數的基因片段。每個基因片段由兩個基因組成。基因分為顯性基因和隱性基因。當一對基因都是顯性基因或者一對基因中一個是顯性基因一個是隱性基因,那麼表現出來的就是顯性性狀;而一對基因都是隱性基因,表現出來的就是隱性性狀。而一般的遺傳病都是隱性性狀,所以遺傳病就是常染色體的陰性形狀表現出來的是遺傳病。

系譜符號
1.由這種致病基因導致的疾病稱為染色體顯性遺傳病。據統計,此類遺傳病或異常性狀已達3711種(1992年)。
致病基因有顯性和隱性之分,其區別在於雜合狀態(Aa)時,是否表現出相應的性或遺傳病。若雜合子(Aa)能表現出與顯性基因A有關的性狀或遺傳病時,其遺傳方式稱為顯性遺傳。
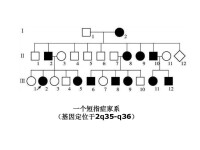
一例短指(趾)症系譜
圖4-2的每個患者基因型都是雜合子(Aa),他(她)們的致病基因A一定來自雙親中的一方,所以雙親中的一方也是Aa,當然也是患者,這樣就出現三代連續傳遞的現象。正常人的基因型都是aa,因此,患者的正常親屬也應都是aa,其子女都可能完全正常。該家系共21人,短指症患者11人(男5女6),發病比例接近1/2。應該指出,這種比例是在大樣本的觀察中方能反映出來,在子女數較少的小家庭往往不能反映出這種特點而出現較大的偏差。上述系譜基本反映了完全顯性遺傳特點,表現在:①連續四代發病;②患者子女中約1/2發病;③男女發病機會大致均等。
(2)不完全顯性:有時雜合子(Aa)的表現型較純合子輕,這種遺傳方式稱為不完全顯性(incompletedominance)或半顯性(semi-dominance),也稱中間型遺傳(intermedeate inheritance)。這裡,雜合子(Aa)中的顯性基因A和隱性基因a的作用都得到-定程度的表達。β地中海貧血可作不完全顯性遺傳實例,致病基因βO純合子,基因型為βOβO者病情嚴重,雜合子基因型為βOβA者病情較輕,而正常基因βA純合子基因型為βAβA者無癥狀。從臨床癥狀輕重來看,雜合子βOβA病情是界於βOβO與βAβA之間(詳見本章第三節)。
(3)共顯性:一對常染色體上的等位基因,彼此間沒有顯性和隱性的區別,在雜合狀態時,兩種基因都能表達,分別獨立地產生基因產物,這種遺傳方式稱為共顯性遺傳(co-dominace)。ABO血型的遺傳可作為顯性遺傳的實例。ABO血型決定於一組復等位基因(multiple alleles)。復等位基因是指在一個群體中,一對特定的基因座位上的基因不是兩種(如A和a),而是三種或三種以上,有時可達數十種。但是,對每一個人來說只能具有其中的任何兩個等位基因。復等位基因是由於一個基因發生多種突變,從而產生多種基因型的結果。ABO血型的基因已定位於9q34,在這一座位上,由IA、IB、和i三種基因組成復等位基因。基因IA對基因i為顯性,基因IB對基因i也是顯性。基因型IAIA和IAi都決定紅細胞膜上抗原A的產生,這種個體為A型血;基因型IBIB和IBi都決定紅細胞膜上抗原B的產生,這種個體為B型血;基因型ii測定H物質的產生而不產生抗原A和抗原B。就IA、IB、i這一組復等位來說,復等位基因的數目是3個,所以共有:n(n+1)/2=3(3+1)/2=6
種基因型。在共顯性時,有4種表現型。如果純合子(IAIA)A型血的人與純合子(IBIB)B型血的人結婚只能出生雜合子(IAIB)AB型血的子女;如果兩個雜合子(IAIB)AB型血的人結婚則會導致1(IAIA):2(IAIB):1(IBIB)的比率,這樣,3:1的比值就被1:2:1的比值所代替,這是兩個等位基因共顯性的結果。
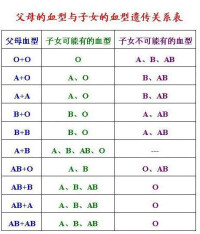
雙親和子女之間血型遺傳的關係
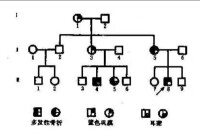
一例成骨不全病例系譜
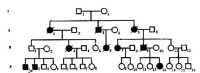
例慢性進行舞蹈病系譜
延遲顯性的一特點是,最年輕一代的患者比例常不足1/2。圖4-4中的第Ⅳ代患者僅3/13即為佐證。

一例白化病患者
苯丙酮尿症
這種先天性代謝病是由於致病基因使人體肝臟內不能形成苯丙氨酸羥化酶(PAH),該酶能促使苯丙氨酸轉化為酪氨酸,由於它的缺乏,導致苯丙氨酸的轉化受阻,造成人體血液和其他組織中苯丙氨酸的積累。過量的苯丙氨酸和它的衍生物——苯丙酮酸就由尿中排出,所以稱之為苯丙酮尿症。苯丙酮酸及其代謝產物如在腦中大量積累,會使腦組織的生化代謝紊亂,阻礙大腦的生長發育,造成智力低下,即是引起該症患者痴獃的原因。另外,過量的苯丙氨酸及其代謝產物可能會抑制酪氨酸向黑色素的轉化,故患者往往伴有膚色和發色較淡的性狀表現。控制苯丙氨酸羥化酶的基因定位於12q22~12q24.2。
黑尿症
人體內酪氨酸的另一條重要代謝途徑是轉化成乙醯乙酸,後者再進一步分解成二氧化碳和水,使得尿液中無尿黑酸存在。但是,當由於基因缺陷造成尿黑酸氧化酶缺乏時,尿黑酸因不能被氧化分解而從尿中排出。尿黑酸本身並無顏色,但在空氣中放置一段時間會變為黑色,於是尿液也隨之變黑。在鹼性條件下能促使尿黑酸更進一步變黑,所以這類患兒的尿布用肥皂洗,就越洗越黑。不過,黑尿症是一種常染色體隱性遺傳的良性病症,一般對患者危害不大,但有時也可使其軟骨和關節等部位產生色素沉積,嚴重時會造成關節炎。
白化病
在正常人體內,酪氨酸還有一條重要代謝途徑,就是在酪氨酸酶的參與下,形成黑色素。黑色素使人的毛髮呈現出黑色,長期從事野外勞動的人,其皮膚也會變得黝黑,這是因為在陽光的照射下,加速了黑色素的形成,使皮膚細胞中有較多的黑色素沉著所致。黑色素的增加可以防止太陽紫外線對人體的照射,有一定的保護作用。但是,如果控制酪氨酸合成酶的基因發生了隱性突變或從雙親那裡得到的是兩個隱性基因,這種人就不能合成酪氨酸酶,從而導致體內黑色素的形成受阻,缺乏黑色素的結果,使人體全身發白,就連頭髮、眉毛也是白的,這就是通常說的白化病,俗稱“羊白頭”。白化病患者的其他方面都正常,不像苯丙酮尿症那樣會影響人的智力發育。這種人的唯一缺陷是怕日光的曝晒,尤其是眼睛特別畏光,平常見到的白化病人幾乎都眯著眼睛,特別嚴重的還可因光線刺激過強而造成失明。
先天性葡萄糖、半乳糖吸收不良症
患兒呈現腹瀉,為水樣便,與尿很相似。腹瀉的發生和程度與餵給糖的時間及量有關,給食后24h出現腹瀉,進食越多越嚴重,但患兒進食量卻又很大,所以體重很快下降。繼而呈現脫水、失重、營養不良。患兒只要不以果糖作為主要糖類來源,就會出現腹瀉,故要終身限制食用葡萄糖及半乳糖。但隨年齡增長對葡萄糖和半乳糖有所耐受。
鐮刀形紅細胞貧血病(sivklecell anemia)
此病常認為是常染色體隱性遺傳,更像不完全顯性,致病基因的純合體貧血嚴重,發育不良,關節、腹部和肌肉疼痛,多在幼年期死亡。但帶有致病基因的雜合體大部分是無癥狀,或有的僅有輕度的貧血。但是,如果這種雜合體的人處於高原地區或長時間進行大強度運動訓練而導致體內缺氧時,紅細胞就會發生“鐮變”,阻塞血管,引起全身發燒,肌肉酸痛,大量紅細胞被脾臟吞噬,血紅蛋白下降,機體運輸O2和CO2的能力降低,造成機體紅細胞破壞和缺氧的惡性循環。據報道,這種雜合體的人在麻醉、輸血、體力消耗等特殊情況下,會出現死亡。如 1970年在美國的德克薩斯州有4個雜合體黑人新兵因應激反應而死亡。
體位性(直)蛋白尿
此病患者腎臟無器質性病變,但長時間立性站立、行走、體力勞動,或進行大運動量訓練時尿蛋白量增多,多系無力型體質。
肝糖原貯積症(glycogen storage disease)
患兒於新生兒期即可出現進行性肝腫大,餵養困難,生長發育遲緩,2歲內易出現嚴重低血糖症,並伴有驚厥。患兒身矮、肌肉鬆弛且發育不良,頰和臀部脂肪組織增多。有乳酸中毒,高脂血症,生長遲滯。肝活檢可見有大量糖原和脂質貯積。
半徠乳糖血症(galactosemia)
正常嬰兒體內的半乳糖經腸道吸收后,在肝內轉變成1-磷酸葡萄糖而被利用。患兒因缺乏1-磷酸半乳糖尿苷轉移酶,進含乳食物后,血中1-磷酸半乳糖及半乳糖濃度明顯升高。1-磷酸半乳糖在肝臟積聚可引起肝腫大,肝功受損;在腦的積聚可引起運動及智力障礙;血中半乳糖升高可使葡萄糖釋出減少,出現低血糖症。另外,晶體內半乳糖增多,激活醛糖還原酶,產生半乳糖醇,引起白內障。轉移酶基因定位在9p13。
丙酮酸激酶缺乏症(pyruvate kinase deficiency)
嬰兒型多在新生兒期即出現癥狀,黃疸與貧血都比較嚴重,黃疸可發生在生后兩天內,甚至需要換血。肝脾明顯腫大,生長、發育受到障礙,重者常需多次輸血才能維持生命。但隨年齡增大,血紅蛋白可以維持在低水平,不再需輸血。化驗可見紅細胞較大,非球型。紅細胞丙酮酸激酶活性降低,常降至正常值的30%左右。此病純合子發病,雜合子不顯癥狀,故患者的父母(攜帶者)丙酮酸激酶活性也輕度降低。成人型癥狀很輕,常被忽視。多於合併感染時才出現貧血。
丙酮酸激酶是紅細胞中的一種酶,其功能是在糖無氧酵解過程中起催化作用。如該酶缺乏可使紅細胞內ATP的產生減少,從而影響紅細胞的壽命,引起溶血性貧血。有三種同工酶:M1、M2、L型, M型基因定位於15q22-qter, L型基因定位於1q21。
黑蒙性痴獃(tay-sachs’s disease)
此病是中樞神經系統有類脂物質積累引起的進行性智力障礙和視力障礙的遺傳性疾病。嬰兒出生后4~8個月間出現癥狀,最初為呆鈍、淡漠,以後坐時頭不能抬直,肌肉無力,體重逐漸降低,已有的運動反射消失,視力減弱以至於失明。眼底檢查黃斑部有櫻桃紅色點,周圍有變性細胞構成的灰白色圈,視神經乳頭蒼白,幾個月後可完全失明,痴獃和不能運動,病情進展,肌肉出現強直痙攣,反射活躍。腱反射亢進、頸肌反射呈陽性,常有驚厥發作。病兒有時狂笑、高叫,在1歲時很胖,最後極度衰竭、消瘦,但肝脾不增大,無嚴重貧血。
患者各種組織中均缺乏氨基己糖酶A,這種酶能裂解神經節苷脂分子末端的N-乙醯半乳糖胺。由於神經節苷脂在腦組織中蓄積,引起一系列臨床癥狀。酶基因定位於 15 q23~q24。
高雪氏病(Gaucher’s disease)
此病因脂代謝紊亂,造成網狀內皮細胞有腦苷脂類沉積,可分急性和慢性兩種。
慢性型:也稱幼兒型 起病較慢,可在任何年齡發病,以學齡前發病較多。起初出現肝腫大及輕度貧血,以後脾腫大,貧血加重,血小板及白細胞明顯減少,可有皮膚、粘膜出血。長骨關節可有隱痛和局限性壓痛,偶可發生病理性骨折,X線檢查可見骨質疏鬆。骨髓、脾、淋巴結穿刺取材塗片,可見高雪氏細胞(直徑 70~80μm、卵圓形、含有一個至數個偏心的細胞核,胞漿淺染呈皺縮樣,無空泡)。
此病患者葡萄糖神經醯胺酶缺乏,神經鞘脂類代謝障礙,葡萄糖腦苷脂蓄積在網狀內皮系統細胞中,主要侵犯肝、脾、淋巴結及骨髓。致病基因定位於1q21。

基本信息
- 中文名
- 常染色體隱性遺傳病
- 外文名
- autosomal recessive disorder
- 致病基因
- 常染色體上
- 基因性狀
- 隱性