戈謝細胞
戈謝細胞
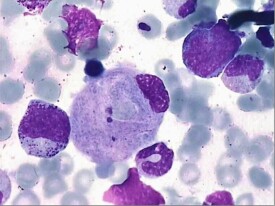
戈謝細胞(Gaucher′s cell),又稱高雪氏細胞,法國皮膚科醫生Gaucher P1882年首先報道,是因溶酶體內的酸性β-葡萄糖苷酶(acid β-glucosidase),又稱葡萄糖腦苷脂酶(glucocerebrosidase,GC)缺陷導致戈謝病(Gaucher′s disease,又稱高雪氏病,徠高雪病),使葡萄糖腦苷脂貯積在各器官的單核巨噬細胞系統中,形成戈謝細胞。戈謝細胞是臨床上診斷戈謝病的重要依據,戈謝病是一種少見的由葡萄糖腦苷脂酶缺陷引起的常染色體隱性遺傳性疾病,該病可在骨髓、肝、脾、淋巴結中找到戈謝細胞。
基本情況介紹
戈謝病
戈謝病病因
發病機制
癥狀
臨床表現
診斷依據
治療前的注意事項
產前診斷
戈謝病西醫療法
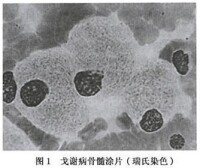
患者骨髓里的戈謝細胞
2.長形類長方形
3.外形不規則
4.近似梭形
5.胞體橢圓形,多個核。
6.胞體橢圓形
7.吞噬型戈謝細胞 漿中有一個淋巴細胞
8.胞漿呈飄帶狀,核居一側,有核仁。
9.巨大戈謝細胞
10.胞體類圓形,雙核對稱分佈於細胞上方兩側,核仁不清。
11.胞體橢圓,為22.7μm×16.311.胞體橢圓,為22.7μm×16.3μm。漿藍,核偏在,核仁不清。
12.具有隱形性
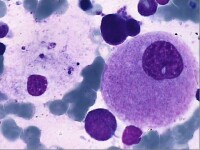
戈謝細胞
1.腦電圖檢查可早發現神經系統浸潤,在神經系統癥狀出現前即有廣泛異常波型。Ⅲ型患者在未出現神經系統癥狀前很難與I型鑒別。通過腦電圖檢查可預言患者將來是否有可能出現神經系統癥狀。
2.產前診斷:患者的母親再次妊娠時可取絨毛或羊水細胞經酶活性測定做產前診斷,若患者的基因型已確定,也可做產前基因診斷。
3.典型的細胞可以在骨髓,脾穿刺或肝活檢的標本中發現。細胞培養無腦苷酯酶的活性則可明確診斷,戈謝病可以通過羊膜穿刺術或絨毛取樣而進行產前診斷、DNA技術可以診斷特定的戈謝病等位基因,編碼葡萄糖苷酸的基因定位於人類染色體的Iq21位。
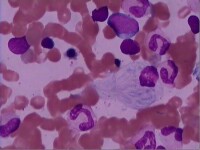
戈謝細胞
溶酶體(lysosome)是一種細胞器,即細胞內的超微結構,為單層包被的囊泡,外面是一層脂蛋白膜。它是細胞的處理與回收系統。內部液體呈酸性,含有60多種酸性水解酶,可降解各種生物大分子,如核酸、蛋白質、脂質、黏多糖及糖原等。組成細胞的各種生物大分子都處於動態平衡中,不斷被分解又不斷被再合成。通過內吞作用攝入的生物大分子也需要分解成不同的組分后,才能被利用。這些大分子的分解都是在溶酶體中進行的。
溶酶體中的每一種酶皆有各自的編碼基因。每一種酶的缺陷直接導致某一特定的生物大分子不能正常降解而在溶酶體中貯積。其共同結果都是溶酶體隨之發生腫脹,細胞也變得臃腫失常,細胞功能受到嚴重影響,最終導致疾病,稱為溶酶體貯積症(lysosomal storage disease,LSD)。
典型的細胞可以在骨髓,脾穿刺或肝活檢的標本中發現。細胞培養無腦苷酯酶的活性則可明確診斷,戈謝病可以通過羊膜穿刺術或絨毛取樣而進行產前診斷。DNA技術可以診斷特定的戈謝病等位基因,編碼葡萄糖苷酸的基因定位於人類染色體的Iq21位。
任何年齡自出生至80歲均可發病,但以少年兒童多發7歲以下更多可分為三型:
為本病最常見類型也是脂質貯積病中常見者。猶太人(Ashkenazi-Jewish民族)中多見,但各民族中均有。在美國估計每年兒童患者不到5000例任何年齡均可起病常以脾臟大就醫進展可快可慢進展慢者脾臟大尤甚有時有脾梗死或脾破裂而發生急腹症癥狀肝臟呈進行性腫大但不如脾臟腫大明顯病程久者,皮膚及黏膜呈茶黃色,常誤診為黃疸,暴露部位如頸、手及小腿最明顯,呈棕黃色眼球結膜上常有楔形瞼裂斑,底在角膜邊緣,尖指向內、外眥,初呈黃白色后變為棕黃色肺累及時可影響氣體交換而出現癥狀。晚期患者四肢可有骨痛甚而病理性骨折,以股骨下端最常見也可累及股骨頸及脊柱骨有脾功能亢進時可因血小板減少而有出血傾向。小兒患者身高及體重常受影響。
患兒自生后即可有肝大脾大3~6個月時已很明顯,有吸吮、吞咽困難生長發育落後表現。神經系統癥狀突出,頸強直頭後仰肌張力增高、角弓反張腱反射亢進,最後變為軟癱,無反應腦神經受累時可有內斜面癱等癥狀。易併發感染。由於病程短暫,多於嬰兒期死亡,因此肝脾臟腫大不如成人型明顯無皮膚色素沉著,骨骼改變不顯著。
常於2歲至青少年期發病脾大常於體檢時發現,一般呈中度腫大,病情發展緩慢,逐漸出現中樞神經系統癥狀,如:肌陣攣性抽搐動作不協調精神錯亂最後卧床不起。肝臟經常輕微腫大但也可進行性腫大而出現肝功能嚴重損害。
戈謝病是一種少見的由葡萄糖腦苷脂酶缺陷引起的常染色體隱性遺傳性疾病,該病可在骨髓、肝、脾、淋巴結中找到戈謝細胞並作為診斷依據之一。新近一些文獻報告,在一些血液病,如急性或慢性粒細胞白血病、多發性骨髓瘤、地中海貧血、先天性紅細胞發育不良貧血等骨髓片。
遺傳性代謝性疾病產前診斷(antenatal diagnosis of hereditary metabolic disease)是防止遺傳病發生的有效措施之一,是人類遺傳學知識的實際應用,是優生的重要措施。本症確定患兒基因型后,其母再次妊娠可做產前基因診斷,也可予雜合子檢查。
羊膜腔穿刺術(amniocentesis)可於妊娠中期17~20周通過腹壁進行,羊水細胞是胎兒脫落的上皮細胞,經培養后可做酶活性測定或基因分析。此方法造成的胎兒丟失率為0.5%。至今仍然是產前診斷的一個重要手段。
絨毛來自胚胎滋養層,可於妊娠10~12周,通過腹壁吸取絨毛。可用於酶活性測定或基因分析。優點是比羊膜腔穿刺提前了2個月,不必培養,可較早獲得產前診斷結果。一旦胎兒患病,孕婦可及時選擇人工流產,後續操作比較容易進行,而且可早日解除孕婦的心理負擔。
產前診斷的先決條件是對先證者做出準確的診斷,母親再次妊娠時才可能在產前診斷時有目的地查某種酶或某種基因檢測。尤其產前基因診斷,除了缺失和用PCR/ASO方法能直接檢測出基因缺陷外,其他連鎖分析方法都以臨床診斷為前提。原因就是某些遺傳病存在遺傳異質性,同樣的疾病表型可由多個基因座突變引起,例如肌營養不良症,較常見的是DMD/BMD,但還有其他基因突變可導致肌營養不良。如果臨床診斷不準確,用A病的多態性位點進行B病的基因診斷,勢必被引入歧途,導致診斷錯誤。其次還要避免樣品污染,胎兒材料中母源DNA的污染不容忽視。血性羊水常是導致診斷錯誤的根源之一,嚴重的血性羊水一定要通過培養去除孕婦的白細胞。絨毛採集后,一定要在倒置顯微鏡下檢查挑選,剔除子宮內膜組織。
在產前清楚判明胎兒是否患病,有的還可在孕早期做出產前診斷,在優生上具有“預防”的意義。因可在臨床上根據明確的產前診斷結果阻止胎兒出生,它不僅是惟一可行的優生措施,而且能減輕家庭及社會的負擔,提高人口素質。
1.一般療法注意營養,預防繼發感染。
2.對症治療 貧血或出血多者可予成分輸血、巨脾或脾功能亢進癥狀明顯者 可考慮切脾,但全脾切除后雖可減輕腹部負擔,減輕貧血和出血傾向,改善發育狀態,偶可自行緩解而痊癒,但有加速肝大及骨骼破壞的可能。故應盡量延遲手術,必要時,可考慮部分脾切除術。骨痛可用腎上腺皮質激素。
3.酶療法 國外近年來採用β-葡糖腦苷脂酶治療本病,取得一定療效。成人型治療1年後一般情況好轉,肝脾明顯縮小,生長發育加快,血紅蛋白升高,血小板亦緩慢上升,肺部受累者,肺功能亦可得到改善。骨病變如舊,但發現治療初期有不伴尿鈣增加的低血鈣情況,推測骨病變好轉可能需較長時間。嬰兒型應用后,肝、脾可縮小,但腦癥狀多不能好轉。目前對應用劑量及方法尚不統一,初步應用結果認為2.3U/kg,每周3次,靜脈滴注,療效與60U/kg,每2周1次療效相似。此提示2周1次的大劑量方法非常不經濟,前種方法可降低極為昂貴的藥費。嬰兒型的劑量一般認為應較大,每月70~120U/kg,每周2或3次。此酶的來源有2種:一為來自胎盤名阿糖腦苷酶(alglucerase)或β-葡萄腦苷酯酶(ceredase),另一為重組品,名imiglucerase或cerezyme,各15例雙盲法比較,療效相仿。
靜脈輸入從胎盤中提煉的葡萄糖腦酯酶可以改善Ⅰ型病人的臨床癥狀,現較通用的方法是每次靜脈輸入超過1~2小時,每2周1次(劑量因人而異,最初的劑量為每次60u/kg).治療計劃(劑量,輸入頻率及輸入速率)還需進一步研究,劑量比FDA提倡的每2周60u/kg略少是較為合適的.
4.骨髓移植異基因骨髓移植治療能使酶活力上升,肝、脾縮小,戈謝細胞減少,但手術危險性與療效必須慎重衡量考慮。
5.基因治療 已試用β-葡糖腦苷脂酶的正常基因插入到自身幹細胞中並進行自身移植,尚需進行繼續研究。
Ⅰ型GD進展緩慢,脾切除后可長期存活,智力正常,惟生長發育落後。GBA替代治療效果顯著,預后最好。Ⅰ型GD脾切除后,肝和骨髓中GC蓄積加快,故可早期死於肺和肝功能障礙,感染出血等。
Ⅱ型GD多於發病後1年內死於繼發感染,少數可存活2年以上。
Ⅲ型GD多由於神經系統癥狀較重,死於病發症。由於GBA的應用,預後有較大的改觀。
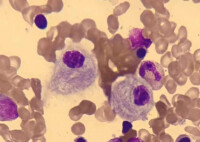
徠戈謝細胞
基本信息
- 中文名
- 戈謝細胞
- 外文名
- Gaucher′s cell
- 別名
- 高雪氏細胞
- 顯狀部位
- 骨髓、肝脾、淋巴結
- 常見病因
- 葡萄糖腦苷脂酶缺陷
- 傳染性
- 無傳染性
- 遺傳性
- 遺傳
- 顯隱性
- 隱性
- 治療途徑
- 酶療法、骨髓移植、基因治療
- 西醫學名
- Gaucher′s cell
- 是否納入醫保
- 否
- 性狀
- 吞噬型、長形、橢圓形