蛋白質結構
蛋白質分子的空間結構
蛋白質結構是指蛋白質分子的空間結構。蛋白質主要由碳、氫、氧、氮等化學元素組成,是一類重要的生物大分子,所有蛋白質都是由20種不同氨基酸連接形成的多聚體,在形成蛋白質后,這些氨基酸又被稱為殘基。
蛋白質和多肽之間的界限並不是很清晰,有人基於發揮功能性作用的結構域所需的殘基數認為,若殘基數少於40,就稱之為多肽或肽。要發揮生物學功能,蛋白質需要正確摺疊為一個特定構型,主要是通過大量的非共價相互作用(如氫鍵,離子鍵,范德華力和疏水作用)來實現;此外,在一些蛋白質(特別是分泌性蛋白質)摺疊中,二硫鍵也起到關鍵作用。為了從分子水平上了解蛋白質的作用機制,常常需要測定蛋白質的三維結構。由研究蛋白質結構而發展起來了結構生物學,採用了包括X射線晶體學、核磁共振等技術來解析蛋白質結構。
一定數量的殘基對於發揮某一生物化學功能是必要的;40-50個殘基通常是一個功能性結構域大小的下限。蛋白質大小的範圍可以從這樣一個下限一直到數千個殘基。估計的蛋白質的平均長度在不同的物種中有所區別,一般約為200-380個殘基,而真核生物的蛋白質平均長度比原核生物長約55%。更大的蛋白質聚合體可以通過許多蛋白質亞基形成;如由數千個肌動蛋白分子聚合形成蛋白纖維。
1959年佩魯茨和肯德魯對血紅蛋白和肌血蛋白進行結構分析,解決了三維空間結構,獲1962年化學獎。
1962年,鮑林發現了蛋白質的基本結構,克里克、沃森在X射線衍射資料的基礎上,提出了DNA三維結構的模型。獲1962年生理或醫學獎。50年代后豪普特曼和卡爾勒建立了應用X射線分析得以直接法測定晶體結構的純數學理論,在晶體研究中具有劃時代的意義,特別在研究大分子生物物質如激素、抗生素、蛋白質及新型藥物分子結構方面趣了重要作用。他們因此獲1985年化學獎。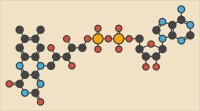
蛋白質結構
蛋白質分子是由氨基酸首尾相連縮合而成的共價多肽鏈,但是天然蛋白質分子並不是走向隨機的鬆散多肽鏈。每一種天然蛋白質都有自己特有的空間結構或稱三維結構,這種三維結構通常被稱為蛋白質的構象,即蛋白質的結構。
蛋白質的分子結構可劃分為四級,以描述其不同的方面:
一級結構:組成蛋白質多肽鏈的線性氨基酸序列。
二級結構:依靠不同氨基酸之間的C=O和N-H基團間的氫鍵形成的穩定結構,主要為α螺旋和β摺疊。
三級結構:通過多個二級結構元素在三維空間的排列所形成的一個蛋白質分子的三維結構。
四級結構:用於描述由不同多肽鏈(亞基)間相互作用形成具有功能的蛋白質複合物分子。
除了這些結構層次,蛋白質可以在多個類似結構中轉換,以行使其生物學功能。對於功能性的結構變化,這些三級或四級結構通常用化學構象進行描述,而相應的結構轉換就被稱為構象變化。
蛋白質的一級結構(primary structure)就是蛋白質多肽鏈中氨基酸殘基的排列順序(sequence),也是蛋白質最基本的結構。它是由基因上遺傳密碼的排列順序所決定的。各種氨基酸按遺傳密碼的順序,通過肽鍵連接起來,成為多肽鏈,故肽鍵是蛋白質結構中的主鍵。
迄今已有約一千種左右蛋白質的一級結構被研究確定,如胰島素,胰核糖核酸酶、胰蛋白酶等。
蛋白質的一級結構決定了蛋白質的二級、三級等高級結構,成百億的天然蛋白質各有其特殊的生物學活性,決定每一種蛋白質的生物學活性的結構特點,首先在於其肽鏈的氨基酸序列,由於組成蛋白質的20種氨基酸各具特殊的側鏈,側鏈基團的理化性質和空間排布各不相同,當它們按照不同的序列關係組合時,就可形成多種多樣的空間結構和不同生物學活性的蛋白質分子。
蛋白質分子的多肽鏈並非呈線形伸展,而是摺疊和盤曲構成特有的比較穩定的空間結構。蛋白質的生物學活性和理化性質主要決定於空間結構的完整,因此僅僅測定蛋白質分子的氨基酸組成和它們的排列順序並不能完全了解蛋白質分子的生物學活性和理化性質。例如球狀蛋白質(多見於血漿中的白蛋白、球蛋白、血紅蛋白和酶等)和纖維狀蛋白質(角蛋白、膠原蛋白、肌凝蛋白、纖維蛋白等),前者溶於水,後者不溶於水,顯而易見,此種性質不能僅用蛋白質的一級結構的氨基酸排列順序來解釋。
蛋白質的空間結構就是指蛋白質的二級、三級和四級結構。
蛋白質的二級結構(secondary structure)是指多肽鏈中主鏈原子的局部空間排布即構象,不涉及側鏈部分的構象。
1.肽鍵平面(或稱醯胺平面,amide plane)。
Pauling等人對一些簡單的肽及氨基酸的醯胺等進行了X線衍射分析,從一個肽鍵的周圍來看,得知:
(1)肽鏈中的C-N鍵長0.132nm,比相鄰的N-C單鍵(0.147nm)短,而較一般C=N雙鍵(0.128nm)長,可見,肽鍵中-C-N-鍵的性質介於單、雙鍵之間,具有部分雙鍵的性質,因而不能旋轉,這就將固定在一個平面之內。
(2)肽鍵的C及N周圍三個鍵角之和均為360°,說明都處於一個平面上,也就是說六個原子基本上同處於一個平面,這就是肽鍵平面。肽鏈中能夠旋轉的只有α碳原子所形成的單鍵,此單鍵的旋轉決定兩個肽鍵平面的位置關係,於是肽鍵平面成為肽鏈盤曲摺疊的基本單位。
(3)肽鍵中的C-N既具有雙鍵性質,就會有順反不同的立體異構,已證實處於反位。
2.蛋白質主鏈構象的結構單元
1)α-螺旋Pauling等人對α-角蛋白(α-keratin)進行了X線衍射分析,從衍射圖中看到有0.5~0.55nm的重複單位,故推測蛋白質分子中有重複性結構,並認為這種重複性結構為α-螺旋(α-helix).
α-螺旋的結構特點如下:
①多個肽鍵平面通過α-碳原子旋轉,相互之間緊密盤曲成穩固的右手螺旋。
②主鏈呈螺旋上升,每3.6個氨基酸殘基上升一圈,相當於0.54nm,這與X線衍射圖符合。
③相鄰兩圈螺旋之間借肽鍵中C=O和H桸形成許多鏈內氫健,即每一個氨基酸殘基中的NH和前面相隔三個殘基的C=O之間形成氫鍵,這是穩定α-螺旋的主要鍵。
④肽鏈中氨基酸側鏈R,分佈在螺旋外側,其形狀、大小及電荷影響α-螺旋的形成。酸性或鹼性氨基酸集中的區域,由於同電荷相斥,不利於α-螺旋形成;較大的R(如苯丙氨酸、色氨酸、異亮氨酸)集中的區域,也妨礙α-螺旋形成;脯氨酸因其α-碳原子位於五元環上,不易扭轉,加之它是亞氨基酸,不易形成氫鍵,故不易形成上述α-螺旋;甘氨酸的R基為H,空間佔位很小,也會影響該處螺旋的穩定。
2)β-片層結構Astbury等人曾對β-角蛋白進行X線衍射分析,發現具有0.7nm的重複單位。如將毛髮α-角蛋白在濕熱條件下拉伸,可拉長到原長二倍,這種α-螺旋的X線衍射圖可改變為與β-角蛋白類似的衍射圖。說明β-角蛋白中的結構和α-螺旋拉長伸展后結構相同。兩段以上的這種摺疊成鋸齒狀的肽鏈,通過氫鍵相連而平行成片層狀的結構稱為β-片層(β-pleated sheet)結構或稱β-折迭。
β-片層結構特點是:
①是肽鏈相當伸展的結構,肽鏈平面之間摺疊成鋸齒狀,相鄰肽鍵平面間呈110°角。氨基酸殘基的R側鏈伸出在鋸齒的上方或下方。
②依靠兩條肽鏈或一條肽鏈內的兩段肽鏈間的C=O與N-H形成氫鍵,使構象穩定。
③兩段肽鏈可以是平行的,也可以是反平行的。即前者兩條鏈從“N端”到“C端”是同方向的,後者是反方向的。β-片層結構的形式十分多樣,正、反平行能相互交替。
④平行的β-片層結構中,兩個殘基的間距為0.65nm;反平行的β-片層結構,則間距為0.7nm.
3)β-轉角
蛋白質分子中,肽鏈經常會出現180°的回折,在這種回折角處的構象就是β-轉角(β-turn或β-bend)。β-轉角中,第一個氨基酸殘基的C=O與第四個殘基的N-H之間形成氫鍵,從而使結構穩定。
4)無規捲曲
沒有確定規律性的部分肽鏈構象,肽鏈中肽鍵平面不規則排列,屬於鬆散的無規捲曲(random coil)。
超二級結構(supersecondary structure)是指在多肽鏈內順序上相互鄰近的二級結構常常在空間摺疊中靠近,彼此相互作用,形成規則的二級結構聚集體。目前發現的超二級結構有三種基本形式:α螺旋組合(αα);β摺疊組合(βββ)和α螺旋β摺疊組合(βαβ),其中以βαβ組合最為常見。它們可直接作為三級結構的“建築塊”或結構域的組成單位,是蛋白質構象中二級結構與三級結構之間的一個層次,故稱超二級結構。
結構域(domain)也是蛋白質構象中二級結構與三級結構之間的一個層次。在較大的蛋白質分子中,由於多肽鏈上相鄰的超二級結構緊密聯繫,形成二個或多個在空間上可以明顯區別它與蛋白質亞基結構的區別。一般每個結構域約由100-200個氨基酸殘基組成,各有獨特的空間構象,並承擔不同的生物學功能。如免疫球蛋白(IgG)由12個結構域組成,其中兩個輕鏈上各有2個,兩個重鏈上各有4個;補體結合部位與抗原結合部位處於不同的結構域。一個蛋白質分子中的幾個結構域有的相同,有的不同;而不同蛋白質分子之間肽鏈中的各結構域也可以相同。如乳酸脫氫酶、3-磷酸甘油醛脫氫酶、蘋果酸脫氫酶等均屬以NAD+為輔酶的脫氫酶類,它們各自由2個不同的結構域組成,但它們與NAD+結合的結構域構象則基本相同。
蛋白質的多肽鏈在各種二級結構的基礎上再進一步盤曲或折迭形成具有一定規律的三維空間結構,稱為蛋白質的三級結構(tertiary structure)。蛋白質三級結構的穩定主要靠次級鍵,包括氫鍵、疏水鍵、鹽鍵以及范德華力(Van der Wasls力)等。這些次級鍵可存在於一級結構序號相隔很遠的氨基酸殘基的R基團之間,因此蛋白質的三級結構主要指氨基酸殘基的側鏈間的結合。次級鍵都是非共價鍵,易受環境中pH、溫度、離子強度等的影響,有變動的可能性。二硫鍵不屬於次級鍵,但在某些肽鏈中能使遠隔的二個肽段聯繫在一起,這對於蛋白質三級結構的穩定上起著重要作用。
現也有認為蛋白質的三級結構是指蛋白質分子主鏈摺疊盤曲形成構象的基礎上,分子中的各個側鏈所形成一定的構象。側鏈構象主要是形成微區(或稱結構域domain)。對球狀蛋白質來說,形成疏水區和親水區。親水區多在蛋白質分子表面,由很多親水側鏈組成。疏水區多在分子內部,由疏水側鏈集中構成,疏水區常形成一些“洞穴”或“口袋”,某些輔基就鑲嵌其中,成為活性部位。
具備三級結構的蛋白質從其外形上看,有的細長(長軸比短軸大10倍以上),屬於纖維狀蛋白質(fibrous protein),如絲心蛋白;有的長短軸相差不多基本上呈球形,屬於球狀蛋白質(globular protein),如血漿清蛋白、球蛋白、肌紅蛋白,球狀蛋白的疏水基多聚集在分子的內部,而親水基則多分佈在分子表面,因而球狀蛋白質是親水的,更重要的是,多肽鏈經過如此盤曲后,可形成某些發揮生物學功能的特定區域,例如酶的活性中心等。
具有二條或二條以上獨立三級結構的多肽鏈組成的蛋白質,其多肽鏈間通過次級鍵相互組合而形成的空間結構稱為蛋白質的四級結構(quarternary structure)。其中,每個具有獨立三級結構的多肽鏈單位稱為亞基(subunit)。四級結構實際上是指亞基的立體排布、相互作用及接觸部位的布局。亞基之間不含共價鍵,亞基間次級鍵的結合比二、三級結構疏鬆,因此在一定的條件下,四級結構的蛋白質可分離為其組成的亞基,而亞基本身構象仍可不變。
一種蛋白質中,亞基結構可以相同,也可不同。如煙草斑紋病毒的外殼蛋白是由2200個相同的亞基形成的多聚體;正常人血紅蛋白A是兩個α亞基與兩個β亞基形成的四聚體;天冬氨酸氨甲醯基轉移酶由六個調節亞基與六個催化亞基組成。有人將具有全套不同亞基的最小單位稱為原聚體(protomer),如一個催化亞基與一個調節亞基結合成天冬氨酸氨甲醯基轉移酶的原聚體。
某些蛋白質分子可進一步聚合成聚合體(polymer)。聚合體中的重複單位稱為單體(monomer),聚合體可按其中所含單體的數量不同而分為二聚體、三聚體……寡聚體(oligomer)和多聚體(polymer)而存在,如胰島素(insulin)在體內可形成二聚體及六聚體。
主條目:四級結構
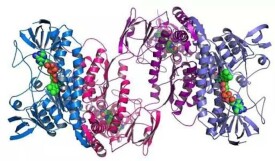
四級結構是由兩個或多個多肽鏈通過相互作用形成的結構。其中,單獨的一條鏈就被稱為亞基。亞基之間不一定要共價連接,但有一些亞基之間是通過二硫鍵來連接的。不是所有的蛋白質都有四級結構,許多蛋白可以以單體形式來發揮功能。四級結構的穩定性與三級結構處於同一水平。兩個或多個亞基形成的複合物統稱為多聚體(multimer),如果是兩個亞基則稱二聚體或二體(dimer),三個亞基稱三聚體或三體(trimer),以此類推。如果多聚體為相同的亞基組成,則加上“同源(homo-)”作為前綴,反之則用“異源(hetero-)”,如同源二聚體或異源三聚體。
植物中的RuBisCo(1,5-二磷酸核酮糖羧化酶/加氧酶)複合物(左)和細菌中的RuBisCo複合物(右)四級結構的比較。前者由12個亞基組成,後者則為2個。
構成生物體內基本物質,為生長及維持生命所必需;
部分蛋白質可作為生物催化劑,即酶和激素;
生物的免疫作用所必需的物資;
有些蛋白質會導致食物過敏。
在結構基因組學中的應用
已經測定了釀酒酵母(Saccharomyces cereuisiae)、線蟲(Caenorhabditis elegans)、果蠅(Drosophilamelanogaster)、擬南芥(Arabidopsis thaliana)等模式生物的基因組序列.。特別值得一提的是,隨著人類基因310福建農林大學學報(自然科學版)第35卷組計劃(Human Genome Program)的完成,接下來的重點就轉移到研究這些基因組裡的所有基因的結構和功能。因此,結構基因組學受到了世界各國的高度重視,美國、日本、歐洲紛紛建立了結構基因組學的研究機構。結構基因組學就是以大規模、高通量測定這些基因的表達產物蛋白質分子的結構為研究目標,以高通量基因克隆技術、蛋白質表達及其純化、蛋白質結晶、蛋白質結構測定為主要研究內容的基因組學分支。
蛋白質結構測定比基因組測定難度大得多,按照常規的實驗步驟,從基因序列到相應的蛋白質結構測定之間還要經過基因表達、蛋白質的提取和純化、結晶、X射線衍射分析等步驟。由於蛋白質結構和性質的多樣性,這些步驟大多沒有固定的規律可循,因而,這種作坊式的需要高超技巧和豐富經驗的研究方法難以適應測定生物蛋白質組中所有蛋白質的要求,因此,需要建立理論分析方法來解決這些問題。以預測技術水平,預測結果的精確度不如X射線衍射分析和NMR等實驗手段,但蛋白質結構預測是大規模、低成本和快速獲得三維結構的有效途徑,例如當目標蛋白質和模板蛋白質的序列相似性超過30%時,以結構預測方法建立的蛋白質三維結構模型就可以用於一般性的功能分析。因而,蛋白質預測技術在結構基因組學中得到了廣泛的應用。
在藥物設計中的應用
從基因組數據到新藥物的過程分為2個部分:一是選擇目標蛋白,二是選擇合適的藥物,藥物分子必需與目標蛋白質分子緊密結合、容易合成且沒有毒副作用。傳統的藥物設計通過篩選大量的天然化合物、已知的底物或配基的類似物(anaIogs)以及生物化學研究來確定前導物(Iead compounds),較少依賴目標蛋白質的三維結構,因而研發周期長、費用巨大,並且帶有或多或少的盲目性。隨著蛋白質結構數據的增長和結構預測技術的發展,目標蛋白質分子三維結構的信息對於上述2個過程發揮著越來越大的作用,計算機輔助的藥物設計(computer-aided drug design)可以縮短研發周期和降低成本。
在蛋白質設計中的應用
蛋白質設計的目標是通過計算機輔助的演演算法以生成符合目標蛋白質三維結構的氨基酸序列,經過漫長的進化,自然界已經篩選出了數量眾多的蛋白質,但天然蛋白質只有在自然條件下才發揮最佳功能,這使得人們利用這些蛋白質受到了限制,因此需要對蛋白質進行改造使其能適應特定條件發揮特定的功能。蛋白質分子的設計分為3類:小改、中改和大改。
化學組成
(1).單純蛋白質:僅含有AAs
(2).結合蛋白質:由AAs和其他非蛋白質化合物所組成
(3).衍生蛋白質:用化學或酶學方法得到的化合物
分子組成
基本單位:氨基酸有不同的AAs通過肽鍵相互連接而成
蛋白質→眎→腖→多肽→二肽→多肽→氨基酸
元素組成
由碳,氫,氧,氮,硫,磷,碘,鐵,鋅等元素組成。
功能分類
(1).結構蛋白質:角蛋白,膠原蛋白,彈性蛋白
(2).有生物活性的蛋白質:酶,激素,免疫球蛋白
(3).食品蛋白質:凡可供食用,易消化,無毒和可供人類利用的蛋白質
主條目:肽鍵兩個氨基酸通過脫水形成肽鍵二面角φ和ψ的圖示。其中黃色部分顯示的是肽平面,而R1和R2分別表示左右兩個殘基的側鏈。
兩個氨基酸可以通過縮合反應結合在一起,並在兩個氨基酸之間形成肽鍵。而不斷地重複這一反應就可以形成一條很長的殘基鏈(即多肽鏈)。這一反應是由核糖體在翻譯進程中所催化的。肽鍵雖然是單鍵,但具有部分的雙鍵性質(由C=O雙鍵中的π電子云與N原子上的未共用電子對發生共振導致),因此C-N鍵(即肽鍵)不能旋轉,從而連接在肽鍵兩端的基團處於一個平面上,這一平面就被稱為肽平面。而對應的肽二面角φ(肽平面繞N-Cα鍵的旋轉角)和ψ(肽平面繞Cα-C1鍵的旋轉角)有一定的取值範圍;一旦所有殘基的二面角確定下來,蛋白質的主鏈構象也就隨之確定。根據每個殘基的φ和ψ來做圖,就可以得到Ramachandran圖,由於形成同一類二級結構的殘基的二面角的值都限定在一定範圍內,因此在Ramachandran圖上就可以大致分辨殘基參與形成哪一類二級結構。下表列出了肽鍵與對應類型單鍵以及氫鍵鍵長的比較。
| 肽鍵 | 平均長度 | 單鍵 | 平均長度 | 氫鍵 | 平均長度(±30) |
| Cα-C | 153pm | C-C | 154pm | O-H---O-H | 280pm |
| C-N | 133pm | C-N | 148pm | N-H---O=C | 290pm |
| N-Ca | 146pm | C-O | 143pm | O-H---O=C | 280pm |
殘基側鏈上的原子根據希臘字母表的順序(α、β、γ、δ、ε等)來命名,如Cα指的是對應殘基上最接近羰基的碳原子,而Cβ則是次接近的。Cα通常被認為是主鏈骨架的組成原子。這些原子之間的鍵對應的二面角則相應以χ1、χ2、χ3等來命名,如賴氨酸側鏈上第一、二個碳原子(即Cα和Cβ)之間共價鍵的二面角為χ1。側鏈可以有多種不同的構象,每一種類型的殘基都有幾種比較穩定的側鏈構象。
許多蛋白質都可以被分為多個結構組成單元,結構域就是這樣一個組成單元。結構域一般可以自穩定,且常常獨立進行摺疊,而不需要蛋白質其他部分的參與;很多結構域都有自己獨特的生物學功能。很多結構域並不是一個基因或基因家族對應蛋白質的獨特結構單元,而往往是許多類蛋白質的共同結構單元。結構域常常是以其生物學功能來命名,如“鈣離子結合結構域”;或以幾類最初發現此結構域的蛋白名稱衍生而來,如PDZ結構域(最初發現於PSD95、DlgA和ZO-1這三個蛋白質)。由於結構域自身可以穩定存在,因此可以將不同來源的結構域通過遺傳工程人為地結合在一起,形成雜合蛋白質。
結構花樣(structural motif)同樣是一種結構組成單元,它是由幾個二級結構的特定組合(如螺旋-轉角-螺旋)所組成;這些組合又被稱為超二級結構。結構花樣往往還包含有長度不同的loop區。
摺疊類型則指的是整體的結構排列類型,如螺旋束和β桶。
儘管真核生物體可以表達數萬種不同的蛋白質,但對應的結構域、結構花樣與摺疊類型的數量卻少得多。一種合理的解釋是,這是進化的結果;因為基因或基因的一部分可以在基因組內被加倍或移動。也就是說,通過基因重組,一個結構域可以從相應蛋白質A移動到本不具有此結構域的蛋白質B上,而其發生的進化驅動力可能是由於該結構域對應的生物學功能趨向於被蛋白質B所利用。
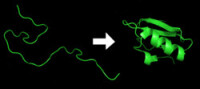
蛋白質摺疊前後
對蛋白質結構進行分類的方法有多種,有多個結構資料庫(包括SCOP、CATH和FSSP)分別採用不同的方法進行結構分類。存放蛋白質結構的PDB資料庫中就引用了SCOP的分類。對於大多數已分類的蛋白質結構來說,SCOP、CATH和FSSP的分類是相同的,但在一些結構中還有所區別。
專門存儲蛋白質和核酸分子結構的蛋白質資料庫中,接近90%的蛋白質結構是用X射線晶體學的方法測定的。X射線晶體學可以通過測定蛋白質分子在晶體中電子密度的空間分佈,在一定解析度下解析蛋白質中所有原子的三維坐標。大約9%的已知蛋白結構是通過核磁共振技術來測定的。該技術還可用於測定蛋白質的二級結構。除了核磁共振以外,還有一些生物化學技術被用於測定二級結構,包括圓二色譜。冷凍電子顯微技術是近年來興起的一種獲得低解析度(低於5埃)蛋白質結構的方法,該方法最大的優點是適用於大型蛋白質複合物(如病毒外殼、核糖體和類澱粉蛋白纖維)的結構測定;並且在一些情況下也可獲得較高解析度的結構,如具有高對稱性的病毒外殼和膜蛋白二維晶體。
| 解析不同解析度的蛋白質結構中可能出現的問題(X射線晶體學) | |
| 解析度(埃) | 結構中可能出現的問題 |
| >4.0 | 單個原子坐標無意義 |
| 3.0-4.0 | 整體摺疊可能是正確的,但很可能有錯誤存在。很多側鏈擺放位置不正確。 |
| 2.5-3.0 | 整體摺疊基本是正確的,除了位於結構表面的一些環狀結構可能沒有正確建模。長側鏈的極性殘基(Lys、Glu、Gln等)和小側鏈殘基(Ser、Val、Thr等)的側鏈擺放位置有可能不正確。 |
| 2.0-2.5 | 與2.5-3.0類似,只是出現錯誤的情況更少。可以明顯觀察到水分子和小配基。 |
| 1.5-2.0 | 側鏈擺放位置基本無誤,甚至一些小的錯誤也可以被檢測到。整體摺疊,包括位於結構表面的環狀結構,基本不可能出現錯誤。 |
| 0.5-1.5 | 在這一解析度下,一般不會有結構錯誤。側鏈異構體庫和立體幾何研究都是利用這一解析度範圍內的結構來進行的。 |
近年來,隨著結構基因組學的興起,大量的蛋白質結構獲得了測定,為研究蛋白質的作用機理提供了重要的結構信息。
測定蛋白質序列比測定蛋白質結構容易得多,而蛋白質結構可以給出比序列多得多的關於其功能機制的信息。因此,許多方法被用於從序列預測結構。
● 二級結構預測
● 三級結構預測
● 同源建模:需要有同源的蛋白三級結構為基礎進行預測。
● Threading法。
● “從頭開始”(Ab initio):只需要蛋白質序列即可進行結構預測。由於運算量大,需要有超級計算機來進行,或採用分散式計算,如Rosetta@home等。
● 四級結構預測:主要是預測蛋白質-蛋白質之間的相互作用方式。
基本信息
- 含義
- 蛋白質分子的空間結構
- 組成元素
- 碳、、氫、氧、氮、硫等化學元素
- 性質
- 一類重要的物生大分子
- 中文名
- 蛋白質結構
- 外文名
- protein structure