共找到2條詞條名為共價鍵的結果 展開
- 化學鍵的一種
- 電子對鍵
共價鍵
化學鍵的一種
共價鍵(covalent bond),是化學鍵的一種,兩個或多個原子共同使用它們的外層電子,在理想情況下達到電子飽和的狀態,由此組成比較穩定的化學結構,像這樣由幾個相鄰原子通過共用電子並與共用電子之間形成的一種強烈作用叫做共價鍵。其本質是原子軌道重疊后,高概率地出現在兩個原子核之間的電子與兩個原子核之間的電性作用。
共價鍵
同一種元素的原子或不同元素的都可以通過共價鍵結合,一般共價鍵結合的產物是分子,在少數情況下也可以形成晶體。
在古希臘,化學還沒有從自然哲學中分離的時代,原子論者對化學鍵有了最原始的設想,恩培多克勒(Empedocles)認為,世界由“氣、水、土、火”這四種元素組成,這四種元素在“愛”和“恨”的作用下分裂並以新的排列重新組合時,物質就發生了質的變化。這種作用力可以被看成是最早的化學鍵思想。
隨後,原子論者德謨克利特設想,原子與原子間,存在著一種“鉤子”,也可以說是粗糙的表面,以致它們在相互碰撞時黏在一起,構成了一個穩定的聚集體。德謨克利特對化學鍵的設想相比於之前的自然哲學家,是更加先進的,他剔除了此類設想中的唯心主義因素。
中世紀的J.R.格勞伯則提出了物質同類相親、異類相斥的思想。其後還出現了關於物質結合的親和力說,認為物質的微粒具有親和力,由此互相吸引而結合在一起。總之,人們關於化學鍵朦朧的認識,啟發了後來的化學家。
18世紀,燃素(phlogiston)的概念進入了化學,並為恩斯特·施塔爾(Ernst Stahl)、亨利·卡文迪許(Henry Cavendish)和約瑟夫·普利斯特里(Joseph Priestley)等先進的化學家所接受。當時,牛頓力學已經提出,他們希望把原子間的作用力和牛頓力學結合起來,給出經典物理學的解釋,但限於當時的條件,這無疑是無法完成的。
1916年,德國化學家阿爾布雷希特·科塞爾(A.Kossel)在考察大量事實后得出結論:任何元素的原子都要使最外層滿足8電子穩定結構,但科塞爾只解釋了離子化合物的形成過程,並沒有解釋共價鍵的形成。
1919年,化學家歐文·朗繆爾首次使用“共價”來描述原子間的成鍵過程
“(原文)we shall denote by the termcovalencethe number of pairs of electrons which a given atom shares with its neighbors”(我們應該用“共價”一詞表示原子間通過共用電子對形成的作用力)
1922年,尼爾斯·玻爾(N.Bohr)從量子化的角度重新審視了盧瑟福的核式模型,這為化學家對化學鍵的認識,提供了全新的平台,他認為電子應該位於確定的軌道之中,並且能夠在不同軌道之間躍遷,定態躍遷可以很好的解釋氫原子光譜的各個譜線。
1923年,美國化學家吉爾伯特·路易斯(G.N.Lewis)發展了科塞爾的理論,提出共價鍵的電子對理論。路易斯假設:在分子中來自於一個原子的一個電子與另一個原子的一個電子以“電子對”的形式形成原子間的化學鍵。這在當時是一個有悖於正統理論的假設,因為庫侖定律表明,兩個電子間是相互排斥的,但路易斯這種
設想很快就為化學界所接受,並導致原子間電子自旋相反假設的提出。
1924年,路易斯·德布羅意(Louis de Broglie)提出波粒二象性的假說,建立了一個原子的數學模型,用來將電子描述為一個三維波形。在數學上不能夠同時得到位置和動量的精確值。
1927年,沃爾特·海特勒(W.H.Heitler)和弗里茨·倫敦(F.London)用量子力學處理氫分子,用近似方法算出了氫分子體系的波函數,首次用量子力學方法解決共價鍵問題。價鍵理論在這一方法的推廣中誕生,他們研究共價鍵的方法就被稱為HL法
1928年,恩利克·費米(Enrica Fermi)提出了一個基於泊松分佈的單電子密度模型試圖解決原子結構問題。之後,道格拉斯·哈特里(Douglas Rayner Hartree)運用迭代法,將體系電子的哈密頓運算元分解為若干個單電子哈密頓運算元的簡單加和,進而將體系多電子波函數表示為單電子波函數的積,改進這一模型,提出哈特里方程。
1930年,哈特里的學生福克(Fock)與約翰·斯萊特(John Clarke Slater)完善了哈特里方程,稱為哈特里-福克方程(HF)。50年代初,斯萊特得到了HF的近似波函數:哈特里-福克-斯萊特方程(HFS)。1963年,赫爾曼(F.Hermann)和斯基爾曼(S.Skillman)把HFS應用於基態原子函數。
1950年,克萊蒙斯·羅瑟恩(C.C.J.Roothaan)進一步提出將方程中的分子軌道用組成分子的原子軌道線性展開,發展出了著名的RHF方程,1964年,計算機化學家恩里克·克萊門蒂(E.Clementi)發表了大量的RHF波函數,該方程以及後續的改進版已經成為現代處理量子化學問題的主要方法。
1929年,貝特等提出配位場理論,最先用於討論過渡金屬離子在晶體場中的能級分裂,後來又與分子軌道理論結合,發展成為現代的配位場理論。
1930年,美國化學家萊納斯·鮑林(L.C.Pauling)在研究碳的正四面體構形時提出軌道雜化理論,認為:能級相近的軌道在受激時可以發生雜化,形成新的簡併軌道,其理論依據就是電子的波粒二象性,而波是可以疊加的。他計算出了多種雜化軌道的形狀,並因在價鍵理論方面的突出貢獻而獲得諾貝爾化學獎。
1932年,弗里德里希·洪德(F.Hund)將共價鍵分為σ鍵、π鍵、δ鍵三種,使價鍵理論進一步系統化,與經典的化合價理論有機地結合起來。
同年,美國化學家羅伯特·馬利肯(Robert S.Mulliken)提出分子軌道理論。認為化合物中的電子不屬於某個原子,而是在整個分子內運動。他的方法和經典化學相距太遠,計算又很繁瑣,一時不被化學界所接受。后經過羅伯特·密立根(Robert A.Millikan)、菲利普·倫納德(Philipp Lenard)、埃里希·休克爾(Erich Hückel)等人的完善,在化學界逐漸得到認可。
1940年,亨利·希吉維克(H.Sidgwick)和托馬斯·坡維爾(Thomas A.Powell)在總結實驗事實的基礎上提出了一種簡單的理論模型,用以預測簡單分子或離子的立體結構。這種理論模型后經羅納德·吉列斯比(R.J.Gillespie)和羅納德·尼霍爾姆(R.S.Nyholm)在20世紀50年代加以發展,定名為價層電子對互斥理論,簡稱VSEPR。VSEPR與軌道雜化理論相結合,可以半定量地推測分子的成鍵方式與分子結構。
1951年,福井謙一提出前線軌道理論,認為,分子中能量最高的分子軌道(HOMO)和沒有被電子佔據的,能量最低的分子軌道(LUMO)是決定一個體系發生化學反應的關鍵,其他能量的分子軌道對於化學反應雖然有影響但是影響很小,可以暫時忽略。HOMO和LUMO便是所謂前線軌道。
1965年,美國化學家羅伯·伍德沃德(Rober B.Woodward)與霍夫曼參照福井謙一的前線軌道理論,提出了分子軌道對稱守恆原理。分子軌道理論得到了新的發展。
由於計算機技術的迅猛發展,和蒙特卡羅方法的應用,量子化學與計算機化學日新月異,對分子結構的推算變得愈發精確期間也誕生了一大批優秀的化學家,據估計,21世紀中期,量子化學還將有新的突破。吉爾伯特·列維斯於1916年最先提出共價鍵。在簡單的原子軌道模型中進入共價鍵的原子互相提供單一的電子形成電子對,這些電子對圍繞進入共價鍵的原子而屬它們共有。
在量子力學中,最早的共價鍵形成的解釋是由電子的複合而構成完整的軌道來解釋的。第一個量子力學的共價鍵模型是1927年提出的,當時人們還只能計算最簡單的共價鍵:氫氣分子的共價鍵。
今天(21世紀初)的計算表明,當原子相互之間的距離非常近時,它們的電子軌道會互相之間相互作用而形成整個分子共享的電子軌道。
化學變化的本質是舊鍵的斷裂和新鍵的形成,化學反應中,共價鍵存在兩種斷裂方式,在化學反應尤其是有機化學中有重要影響。
共價鍵在發生均裂時,成鍵電子平均分給兩個原子(團),均裂產生的帶單電子的原子(團)稱為自由基,用“R·”表示,自由基具有反應活性,能參與化學反應,自由基反應一般在光或熱的作用下進行。
由共價鍵異裂引發的反應稱離子型反應,其下又可分為兩種
·親電反應(electrophilic reaction)
·親核反應(nucleophilic reaction)
離子型反應一般在酸鹼或極性物質的催化下進行。
主條目:路易斯理論
路易斯理論,又稱“八隅體規則”、“電子配對理論”是最早提出的,具有劃時代意義的共價鍵理論,它沒有量子力學基礎,但因為簡單易懂,也能解釋大部分共價鍵的形成,至今依然出現在中學課本里。
共用電子對理論有以下幾點
·原子最外層達到8電子時是穩定結構,化合物中的所有原子的最外層價電子數必須為8(氫為2);
·原子間形成共價鍵時,可通過共用電子的方式使最外層達到8(2)電子穩定結構。
路易斯理論的電子配對思想為價鍵理論的發展奠定了基礎。值得注意的是,路易斯理論尚不完善,它無法說明電子配對的原因和實質;此外,不符合“八隅體規則”的化合物也有很多,例如:三氟化硼(6電子)、五氯化磷(10電子)、六氟化硫(12電子)。
價鍵理論是基於路易斯理論電子配對思想發展起來的共價鍵理論。價鍵理論將應用量子力學解決氫分子問題的成果推廣到其他共價化合物中,成功解釋了許多分子的結構問題。
海特勒-倫敦法
沃爾特·海特勒(W.H.Heitler)和弗里茨·倫敦(F.London)在運用量子力學方法處理氫氣分子的過程中,得到了分子能量E和核間距R之間的關係曲線,發現:若兩個氫原子自旋方向相反,隨著軌道的重疊(波函數相加)會出現一個概率密度較大的區域,氫原子將在系統能量最低核間距處成鍵;若兩個氫原子自旋方向相同,則相減的波函數單調遞減,系統能量無限趨近E=0,沒有最低點,無法成健。因此,價鍵理論通過對氫分子的研究闡明了電子配對的內在原因和共價鍵的本質,價鍵理論就在HL的推廣中誕生。
軌道雜化理論
價鍵理論在解釋分子中各原子分佈情況時,萊納斯·鮑林(L.Pauling)提出了軌道雜化理論。
理論要點有
·中心原子能量相近的不同軌道在外界的影響下會發生雜化,形成新的軌道,稱雜化原子軌道,簡稱雜化軌道;
·雜化軌道在角度分佈上,比單純的原子軌道更為集中,因而重疊程度也更大,更加利於成鍵;
·參加雜化的原子軌道數目與形成的雜化軌道數目相等,不同類型的雜化軌道,其空間取向不同。
註:此為雜化軌道的空間取向,不是化合物的結構,在化合物中,這些軌道可能被孤對電子填充,例如,N原子進行sp^2雜化形成的NO2分子中,有一對孤對電子,那麼NO2的空間結構是折線形(正三角形的一個頂點是孤對電子,電子是“看不見”的)。
(VSEPR theory)
價層電子對互斥理論是一個用來預測單個共價分子形態的化學模型。理論通過計算中心原子的價層電子數和配位數來預測分子的幾何構型,其理論要點有
·共價分子中,中心原子周圍電子對排布的幾何形狀,主要決定於中心原子的價電子層中的電子對數(包括成鍵電子對和孤對電子)。這些電子的位置傾向於分離的儘可能遠一些,使彼此受到的排斥力最小;
·電子層中電子對相互排斥作用的大小,取決於電子對間的相互角度和電子對的成鍵情況。相距角度小,排斥力大。成鍵電子對因受兩個原子吸引,電子云較為緊縮,對其相鄰電子對的斥力小於僅受一個原子核吸引的孤對電子對其相鄰電子對的斥力。即,電子對間斥力大小順序為:孤對電子-孤對電子>孤對電子-成鍵電子對>成鍵電子對-成鍵電子對;
·分子中的雙鍵、三鍵當作單鍵處理;
推測分子構形
設中心原子為A,其餘n個配位原子均用B表示,m對孤對電子用E表示,則該物質可表示為ABnEm。令z=n+m,B和E都用Y表示,則該物質可表示為AYz,這裡的Y就表示中心原子的價電子層中的電子對,z就表示中心原子的價電子層中的電子對數。我們可根據如下公式推測分子構型:
n由化學式即可看出
m=1/2(中心原子價電子數-配位原子提供的電子總數±離子電荷數)
z=n+m
註:更詳細的表參見wikipedia,VSEPR theory(擴展閱讀)
舉例:推測CO3-2的分子構型
n=3
z=3+0=3
故CO3-2的分子構型為平面三角形
分子軌道理論是比價鍵理論更精確的方法,其理論要點有
·分子中的電子不屬於某個原子軌道,而屬於整個分子;
·分子軌道由原子軌道線性組合而成,分子軌道數目等於組成分子軌道的原子軌道數目,其中些軌道能量降低,成為“成鍵軌道”另一些能量升高,成為“反鍵軌道”,還有一些能量不變,稱“非鍵軌道”;
·原子軌道在線性組合時,遵守“對稱性匹配原則”、“能量相近原則”、“最大重疊原則”;
·電子在分子軌道中排布時,遵守“能量最低原理”、“泡利不相容原理”、“洪特規則”;
分子軌道理論能解釋一些價鍵理論無法解釋的現象,比如氧分子的順磁性。
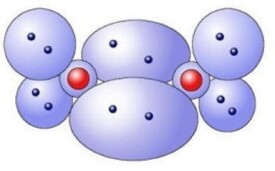
氧氣分子
氧原子的外層電子數為6,這六個電子中的四個組成兩對,其它兩個單獨存在。
每個氧原子有六個外層電子
這兩個單獨的電子與另一個原子中相應的單獨的電子結合組成兩個新的共用的電子對,由此達到電子飽和的狀態。
需要說明的是這裡所描述的氧分子的模型是一個簡化了的模型,實際上的氧分子要比這裡描述的要複雜得多,因為這6個外層原子分佈在不同的軌道上,因此它們不能形成這樣簡單的電子對。實際上的氧分子有三對共用的電子對和兩個單獨的電子。
共價鍵從不同的角度可以進行不同的分類,每一種分類都包括了所有的共價鍵(只是分類角度不同)。
σ鍵
σ鍵(sigma bond)
由兩個原子軌道沿軌道對稱軸方向相互重疊導致電子在核間出現概率增大而形成的共價鍵,叫做σ鍵,可以簡記為“頭碰頭”(見右圖)。σ鍵屬於定域鍵,它可以是一般共價鍵,也可以是配位共價鍵。一般的單鍵都是σ鍵。原子軌道發生雜化后形成的共價鍵也是σ鍵。由於σ鍵是沿軌道對稱軸方向形成的,軌道間重疊程度大,所以,通常σ鍵的鍵能比較大,不易斷裂,而且,由於有效重疊只有一次,所以兩個原子間至多只能形成一條σ鍵。
π鍵(pi bond)
π鍵
成鍵原子的未雜化p軌道,通過平行、側面重疊而形成的共價鍵,叫做π鍵,可簡記為“肩並肩”(見右圖)。π鍵與σ鍵不同,它的成鍵軌道必須是未成對的p軌道。π鍵性質各異,有兩中心,兩電子的定域鍵,也可以是共軛Π鍵和反饋Π鍵。兩個原子間可以形成最多2條π鍵,例如,碳碳雙鍵中,存在一條σ鍵,一條π鍵,而碳碳三鍵中,存在一條σ鍵,兩條π鍵。
π鍵中的π電子可以吸收紫外線並被激發,所以,含有π鍵的化合物有抵禦紫外線的功能,防晒霜正是利用了這個原理防護紫外線對人的傷害。
苯分子中的大π鍵
共軛π鍵具有特殊的穩定性,例如苯環中存在6中心6電子的大π鍵,顯現出芳香性,不易發生加成和氧化反應,而易發生親電取代,與苯環有類似鍵型的化合物包括部分雜環化合物、稠環烴和其他烴類,化學家埃里希·休克爾通過分子軌道計算得出了環烯烴芳香性判定的休克爾規則(亦名4n+2規則),其它常見的非苯芳烴包括薁、輪烯等;而石墨的每一層都有一個無窮大的π鍵,電子在這個超大π鍵中可以自由移動,類似於金屬鍵,這也是石墨可以橫嚮導電的原因。
δ鍵(delta bond)
δ鍵
由兩個d軌道四重交蓋而形成的共價鍵稱為δ鍵,可簡記為“面對面”(見下圖)。
δ鍵只有兩個節面(電子云密度為零的平面)。從鍵軸看去,δ鍵的軌道對稱性與d軌道的沒有區別,而希臘字母δ也正來源於d軌道。
δ鍵常出現在有機金屬化合物中,尤其是釕、鉬和錸所形成的化合物。通常所說的“四重鍵”指的就是一個σ鍵、兩個π鍵和一個δ鍵。
其他
以上三種化學鍵經過組合,可以形成各種不同的鍵型,例如,一個σ鍵和兩個π鍵可以組成一個三鍵,但,有證據表明雙原子間的共價鍵最多不能超過六條。
一般共價鍵
一般共價鍵有時也稱“正常共價鍵”,是為了和“配位共價鍵”進行區分時使用的概念,指成鍵時兩個原子各自提供一個未成對電子形成的共價鍵。
配位共價鍵(coordinate covalent bond)
配位共價鍵簡稱“配位鍵”是指兩原子的成鍵電子全部由一個原子提供所形成的共價鍵,其中,提供所有成鍵電子的稱“配位體(簡稱配體)”、提供空軌道接納電子的稱“受體”。常見的配體有:氨氣(氮原子)、一氧化碳(碳原子)、氰根離子(碳原子)、水(氧原子)、氫氧根(氧原子);受體是多種多樣的:有氫離子、以三氟化硼(硼原子)為代表的缺電子化合物、還有大量過渡金屬元素。對配位化合物的研究已經發展為一門專門的學科,配位化學。
主條目:路易斯酸、路易斯鹼
從上面的內容可以看出,“氫氧根”屬於配體、而“氫離子”屬於受體,這表明,氫離子與氫氧根發生的酸鹼中和反應可以看成是氫離子與氫氧根形成配位鍵的過程。化學家路易斯從這一點出發,提出了“路易斯酸”與“路易斯鹼”的概念,認為凡是在配位鍵成鍵過程中,能給出電子的,都稱為“鹼”;能接納電子的,都稱為“酸”。路易斯的酸鹼理論把酸和鹼的範圍擴大了,路易斯酸鹼對不僅包括所有的的阿倫尼烏斯酸鹼對,還包括一些中性甚至是根本不溶於水的物質。其實,路易斯酸的本質是配位鍵中的“受體”;路易斯鹼的本質是配位鍵中的“配體”,二者是等同的。
配位共價鍵與一般共價鍵的異同
配位共價鍵與一般共價鍵的區別只體現在成鍵過程上,它們的鍵參數是相同的,例如,銨根離子的氮氫鍵中,有三條是一般共價鍵,一條是配位共價鍵,但這四條鍵完全等價,銨根離子也是完全對稱的正四面體形。在書寫時,一般共價鍵使用符號“—”;配位共價鍵使用符號“→”箭頭從配體指向受體。
按成鍵電子偏向極性共價鍵(polar bond)
極性鍵,標&
在化合物分子中,不同種原子形成的共價鍵,由於兩個原子吸引電子的能力不同,電子云偏向吸引電子能力較強的原子一方,因而吸引電子能力較弱的原子一方相對的顯正電性。這樣的共價鍵叫做極性共價鍵,簡稱極性鍵。形成共價鍵時,由於電子云的偏離程度不同,極性鍵又有“強極性鍵”和“弱極性鍵”之分,但通常兩個不同原子間的成鍵就是極性鍵。共價鍵的極性可用鍵矩進行判斷。共價分子的極性等於分子中所有共價鍵偶極矩的矢量和,所以,由極性共價鍵組成的分子可以是極性分子(氯化氫)也可以是非極性分子(二氧化碳)。
非極性共價鍵(non-polar bond)
由同種元素的原子間形成的共價鍵,叫做非極性共價鍵。同種原子吸引共用電子對的能力相等,成鍵電子對勻稱地分佈在兩核之間,不偏向任何一個原子,成鍵的原子都不顯電性。非極性共價鍵存在於單質中,也存在於某些化合物中,完全由非極性鍵構成的分子一定是非極性分子(但有的非極性分子中含有極性鍵)。
共價鍵是電子云的重疊,所以共價鍵最本質的分類方式就是它們的重疊方式。現在已知有3種重疊方式,分別稱作:
σ鍵π鍵δ鍵在有機化合物中,通常把共價鍵以其共用的電子對數分為單鍵、雙鍵以及三鍵。單鍵是一根σ鍵;雙鍵和三鍵都含一根σ鍵,其餘1根或2根是π鍵。
但無機化合物不用此法。原因是,無機化合物中經常出現的共軛體系(離域π鍵)使得某兩個原子之間共用的電子對數很難確定,因此無機物中常取平均鍵級,作為鍵能的粗略標準。
假如組成共價鍵的原子的電負性不同,那麼它們共用的電子對可能被其中的一個原子核吸引,由此而來它們在分子中的分佈也不相等,電子被吸引比較集中的地方顯負性,電子比較少集中的地方顯正性。這樣整個分子就會顯示出一定的極性。一個分子的電極的分佈除其原子的電負性外還與其分子的組成有關。
主條目:配位鍵
配位鍵是一種特殊的共價鍵,它的特點在於共用的一對電子出自同一原子。形成配位鍵的條件是,一個原子有孤電子對,而另一個原子有空軌道。配位化合物,尤其是過渡金屬配合物,種類繁多,用途廣泛,現已形成配位化學。
中心離子:在配合物中,提供空軌道的一方稱為中心離子
配體:在配合物中,提供孤對電子的一方稱為配體
分類化學鍵
共價鍵
σ鍵:三中心兩電子鍵(香蕉鍵)·三中心四電子鍵(氫鍵、雙氫鍵、抓氫鍵)·四中心兩電子鍵
π鍵:反饋π鍵·共軛·超共軛效應·方向性
δ鍵:四重鍵·五重鍵·六重鍵
氫鍵雙氫鍵·雙氫配合物·低能壘氫鍵·對稱氫鍵·親水性
非共價鍵范德華力·機械結合作用·嵌入·鹵鍵·親金作用·重疊·熵力·極性
其他分子內作用力·分子間作用力·配位鍵·齒合度·離子鍵·金屬鍵·成鍵·反鍵·二硫鍵·肽鍵·磷酸二酯鍵
非金屬單質(H2、O2)、共價化合物(NH3、CH4、H2O)、離子化合物(NaOH、NH4Cl)。
①電負性相同或相差很小的非金屬元素原子之間形成共價鍵。
②一般成鍵原子有未成對電子(自旋相反)。
③成鍵原子的原子軌道在空間重疊。
高概率地出現在兩個原子核之間的電子與兩個原子核之間的電性作用。
共價鍵的性質可以通過稱為鍵參數的某些物理量來描述:
鍵級
鍵能
鍵長
鍵角
鍵矩
根據原子軌道最大重疊原理,成鍵時軌道之間可有兩種不同的重疊方式,從而形成兩種類型的共價鍵——σ鍵和π鍵。共價鍵是電子云的重疊,所以共價鍵最本質的分類方式就是它們的重疊方式。現在(21世紀初)已知有3種重疊方式,分別稱作:在有機化合物中,通常把共價鍵以其共享的電子對數分為單鍵、雙鍵以及三鍵。單鍵是一根σ鍵;雙鍵和三鍵都含一根σ鍵,其餘1根或2根是π鍵。但無機化合物不用此法。原因是,無機化合物中經常出現的共軛體系(離域π鍵)使得某兩個原子之間共享的電子對數很難確定,因此無機物中常取平均鍵級,作為鍵能的粗略標準。
(1)σ鍵:以“頭碰頭”方式進行重疊,軌道的重疊部分沿鍵軸呈圓柱形對稱分佈,原子軌道間以重疊方式形成的共價鍵。如:
①H2分子的s-sσ鍵
②HCl分子的s-pσ鍵
③Cl2分子的p-pσ鍵
(2)π鍵:p電子和p電子除能形成σ鍵外,還能以“肩並肩”的方式進行重疊形成π鍵。每個π鍵的電子云由兩塊組成,分別位於由兩原子核構成平面的兩側,如果以它們之間包含原子核的平面鏡面,它們互為鏡像,這種特徵稱為鏡像對稱。
在共價鍵的形成過程中,一個原子中的一個未成對電子與另一個原子中的一個未成對電子配成鍵后,一般來說就不能再與其他原子的未成對電子配成鍵,即每個原子所能形成共價鍵的數目或以單鍵連接的原子數目是一定的,飽和性決定了原子形成分子時相互結合的數量關係。共價鍵的飽和性決定了各種原子形成分子時相互結合的數量關係],是定比定律(law of definite proportion)的內在原因之一。
形成共價鍵時,原子軌道重疊愈多,電子在核間出現的概率愈大,所形成的共價鍵就愈牢固,因此共價鍵將儘可能地沿著電子概率出現最大的方向形成,這就是共價鍵的方向性。影響共價鍵的方向性的因素為軌道伸展方。
在元素符號周圍用小黑點·或×來描述分子中原子共用電子以及未成鍵的價電子的式子,叫電子式。
在共價鍵中,被共享的電子被所有進入共價鍵的原子吸引,由此使得這些原子結合在一起。雖然其原子核之間和電子之間由於電荷互相排斥,但這些排斥作用被位於原子核間的電子減弱,而電子與原子核之間的相互作用更加強。
基本信息
- 中文名
- 共價鍵
- 外文名
- covalent bond
- 分類
- σ鍵,π鍵,配位鍵等
- 類別
- 化學鍵
- 本質
- 原子間的靜電作用