圖位克隆法
圖位克隆法
圖位克隆法(Map-basedcloning)是新型分離和克隆植物基因的方法之一,在不知道基因的表達產物,在未知基因的功能信息又無適宜的相對錶型用於表型克隆時,最常用的基因克隆技術就是圖位克隆法。圖位克隆是通過獲析突變位點與已知分子標記的連鎖關係來確定突變表型的遺傳基本。圖位克隆法隨著相關配套技術(序列資料庫、分子標記等)的日漸成熟,應用範圍將越來越廣。
圖位克隆(Map-based cloning)又稱定位克隆(positional cloning),1986年首先由劍橋大學的Alan Coulson提出,用該方法分離基因是根據目的基因在染色體上的位置進行基因克隆的一種方法,在利用分子標記技術對目的基因進行精確定位的基礎上,使用與目的基因緊密連鎖的分子標記篩選DNA文庫,從而構建目的基因區域的物理圖譜,再利用此物理圖譜通過染色體步行逼近目的基因或通過染色體登陸的方法最終找到包含該目的基因的克隆,最後通過遺傳轉化和功能互補驗證最終確定目的基因的鹼基序列。
圖位克隆的特點是無需預先知道基因的DNA順序,也無需預先知道其表達產物的有關信息,但應有以下兩方面的基本情況。一是有一個根據目的基因的有無建立起來的遺傳分離群體,如F2、DH、BC、RI等。二是開展以下幾項工作:
1)首先找到與目標基因緊密連鎖的分子標記;
2)用遺傳作圖和物理作圖將目標基因定位在染色體的特定位置;
3)構建含有大插入片段的基因組文庫(BAC或YAC庫);
4)以與目標基因連鎖的分子標記為探針篩選基因組文庫;
5)用獲得陽性克隆構建目的基因區域的跨疊群;
6)通過染色體步行、登陸或跳躍獲得含有目標基因的大片段克隆;
7)通過亞克隆獲得帶有目的基因的小片段克隆;
8)通過遺傳轉化和功能互補驗證最終確定目標基因的鹼基序列。
下面將對其中幾個關鍵環節予以敘述:
1.1構建遺傳作圖群體
用於作圖群體的類型可有多種。一般說來,在這類群體中,異花授粉植物分子標記的檢出率較自花授粉植物的高。但是對於基因的圖位克隆而言,培育特殊的遺傳群體是篩選與目標基因緊密連鎖的分子標記的關鍵環節。這些遺傳材料應該滿足這樣的條件,即除了目標基因所在座位的局部區域外,基因組DNA序列的其餘部分都是相同的,在這樣的材料間找到的多態性標記才可能與目標基因緊密連鎖。
目標基因的近等基因系(NILs)是符合條件的一類群體。近等基因系是指幾乎僅在目標性狀上存在差異的兩種基因型個體,這可通過連續回交的途徑獲得。由於NILs的遺傳組成特點,一般凡是能在近等基因系間揭示多態性的分子標記就極有可能位於目標基因的兩翼附近。Martin等(1993)就是用RAPD技術分析番茄PTO基因的近等基因系而獲得了與該基因表現共分離的分子標記,以該分子標記為探針篩選基因組文庫而實現了染色體登陸。又如在玉米上,利用AFLP技術分析玉米CMS-S型雄性不育的恢復基因及近等基因系,已獲得與目標基因緊密連鎖的分子標記並以其用作分離RF3基因的探針。
一般說來,當標記為顯性遺傳時,欲獲得最大遺傳信息量的F2群體,須藉助於進一步的子代測驗,以分辨F2中的雜合體。為此,Michelmore等(1991)發明了分離群體分組分析法(bulked segregant analysis,BSA)以篩選目標基因所在局部區域的分子標記。其原理是將目標基因的F2(或BC1)代分離體的各個體僅以目標基因所控制的性狀按雙親的表型分為兩群,每一群中的各個體DNA等量混合,形成兩個DNA混合池(如抗病和感病、不育和可育)。由於分組時僅對目標性狀進行選擇,因此兩個DNA混合池之間理論上主要在目標基因所在局部區域的差異,這非常類似於NILs,故也稱作近等基因池法。研究表明,近等基因系法、分離群體分組分析法再結合AFLP等強有力的分子標記技術使人們能夠在短時間內從數量眾多的分子標記中篩選出與目標基因緊密連鎖的標記。更為有意義的是,這種策略在尚未構建遺傳圖譜的物種中也是可行的。
1.2篩選與目標基因連鎖的分子標記
篩選與目的基因連鎖的分子標記是圖位克隆技術的關鍵,常用的分子標記有RFLP、RAPD、SSR和AFLP等。這些技術有別於最初的形態標記、細胞學標記和生化標記,一般表現出穩定、可靠的特點,有的使用成本也相對較低,特別適用於篩選與目標基因緊密連鎖的標記,尤其是與目標基因共分離的標記。因此能夠加快克隆基因的進程,研究者可依據不同的研究目標、對象、目的、條件等,選擇使用這些技術。值得提及的是隨著分子標記技術的發展,一些植物的遺傳圖譜構建和比較基因組的研究也有了長足的進步,它們相得益彰,互為促進,為基因的圖位克隆提供了有益的借鑒。
1.3藉助連鎖圖譜篩選分子標記
當基因圖位克隆策略剛剛提出的時候,植物分子連鎖圖譜的構建尚處於萌芽階段,當時找到一個與目標基因連鎖的分子標記通常要花費數月甚至數年的時間。在過去的十年裡,這種狀況有了顯著改善,由於可以很方便的建立和維持建圖所必需的較大的分離群體,因而植物分子連鎖圖構建工作的發展速度超過了動物的同類研究。現在業已建圖的植物已多達幾十種,其中包括了所有重要的農作物,水稻,玉米,番茄,小麥,馬鈴薯,擬南芥等重要經濟作物和模式植物的遺傳圖譜已相當精細,含有數百甚至數千個標記。高密度分子連鎖圖的繪製為篩選與目的基因緊密連鎖的分子標記提供了良好的開端。比如,番茄的遺傳圖譜含有1000個分子標記,其基因組大小約為1000×106bp,因此對任何基因,都可以找到與之相距在1000kb之內的分子標記。
1.4藉助比較基因組共享分子標記
比較基因組研究主要是利用相同的DNA分子標記(主要是cDNA標記和基因克隆)在相關植物種之間進行遺傳或物理作圖,比較這些標記在不同物種基因組中的分佈特點,揭示染色體或染色體片段上的基因及其排列順序的相同(共線性)或相似性(同線性),並由此對相關物種的基因組結構和起源進化進行分析。已有的研究表明,存在生殖隔離的不同植物物種之間在標記探針的同源性、拷貝數及連鎖順序上都具有很大程度的保守性。如番茄、馬鈴薯和辣椒;水稻、小麥和玉米;小麥、黑麥和大麥;高粱和玉米;擬南芥和芸薹屬;以及大麥和水稻。更為有趣的是,Moore等(1995)同時對水稻、小麥、玉米、穀子、甘蔗及高粱等6種主要禾本科物種的基因組進行了比較作圖研究,結果表明其中基因組最小的水稻居於中樞的位置,即這些禾本科植物基因組的保守性可歸結到水稻基因組的19個連鎖區段上。由這19個區段可實現對所研究的全部禾穀類作物染色體的重建,並可構成一個禾穀類作物祖先種的染色體骨架。通過這一研究,人們對禾穀類作物的進化演變歷程有了更深入的認識。植物比較基因組的研究成果表明,在更廣的範圍上,包括禾本科、十字花科、豆科植物及一些樹木的基因組在基因組成和基因排列順序上都存在很大程度的一致性。而在包括小麥、玉米和水稻等重要農作物的禾本科作物上,基因組成和排列順序的一致性非常完好,人們可以在模式植物水稻上用圖位克隆技術克隆相關物種的大多數重要基因。此外,比較基因組研究還給人們一個重要的啟示,那就是模式植物上遺傳作圖的成果可推而廣之,這對那些遺傳作圖工作相對滯后的植物尤其重要。比較基因組作圖使建立高密度遺傳連鎖圖可資利用的標記顯著增加,從而大大提高獲取離目標基因很近的分子標記的可能性。
比較基因組作圖是利用分子標記技術在1個目標性狀的分離群體中把目的基因定位於染色體的一定區域內,然後通過利用高密度的分子連鎖分析,以精細定位目的基因。目前,作圖的類型可分為2類,分別是遺傳圖譜和物理圖譜。遺傳圖譜對圖位克隆雖然非常重要,但更精細的物理圖譜尤為重要。而增加圖譜上的分子標記數目是精細作圖的基礎,增加分子標記的方法—是整合已有的遺傳圖譜,再者就是尋找新的分子標記。現在已有越來越多的植物的遺傳圖譜趨於飽和,如水稻已有3275個分子標記的圖譜,覆蓋基因組的1512.5cM和10400個EST標記。目前利用稀有切點內切酶結合PEGE技術已成功用於植物基因組的大規模物理作圖,從而可以確定連鎖標記與目的基因間的實際物理距離的大小,為基因的克隆奠定了基礎。
1.5局部區域的精細定位、作圖
一旦把目標基因定位在某染色體的特定區域后,接下來就要對其進行精細的作圖及篩選與目標基因緊密連鎖的分子標記。目前的作圖類型分為兩類,分別是遺傳圖譜和物理圖譜。
(1)遺傳圖譜的構建
染色體的交換與重組是遺傳圖譜構建的理論基礎,通過作圖群體的分析,如果一個基因與兩側最近的分子標記距離均在2cM以上,就需要作精細的遺傳圖譜。因為即使是在小基因組水稻中,平均1cM也相當於250kb左右的物理距離,如果在著絲粒附近就可能相當於1000kb左右,需要進行若干步的染色體步行,非常費時費力。因此,精細的遺傳圖譜對圖位克隆顯得非常重要,而增加遺傳圖譜上的分子標記數目是精細作圖的基礎。在一個已知區域內增加分子標記有兩種方法,第一種方法是整合已有的遺傳圖譜。如將生理、生化、分子標記圖在同一區域內整合在一起,就會提高分子標記的密度。另一種方法是尋找新的分子標記。在植物上,精細作圖的發展速度超過了動物的同類研究,這主要是因為在植物上可很方便地建立和維持較大的分離群體,並建立了幾種不同的檢測標記間連鎖關係的統計軟體。現在,業已建圖的植物已多達幾十種,其中包括了所有重要的農作物(Tanksley,1995),如玉米、番茄、水稻、小麥、大麥、燕麥、大豆、高粱、油菜、萵苣、馬鈴薯等。尤其值得一提的是,過去被公認為難以開展遺傳作圖的木本植物,如今也湧現出了許多密度可觀的分子標記連鎖圖,如蘋果和松樹等。由於許多科學家的共同努力和連年的工作積累,已有越來越多生物的遺傳圖譜趨於飽和,這就為生物的物理圖譜的構建奠定了基礎。
(2)物理圖譜的構建
由於分子標記與目標基因之間的實際距離是按鹼基數(bp)來計算的,它會因不同染色體區域基因重組值不同而造成與遺傳距離的差別,所以物理圖譜是真正意義上的基因圖譜。物理圖譜的種類很多,從簡單的染色體分帶圖到精細的鹼基全序列都是物理圖譜。最為常用的物理圖譜有限制酶切圖譜、跨疊克隆群和DNA序列圖譜等。限制酶切圖譜是用幾種限制性內切酶消化DNA,通過電泳檢查限制性片段長度的辦法確定它們的排列順序;對於較大的基因組,可以利用稀有切點限制性內切酶和脈衝電脈。製作跨疊克隆群則需具有一定容量的大片段基因組文庫。比較各個克隆的插人片段,將它們排列成與原來在染色體中的順序一樣的連續克隆群,即跨疊克隆群(也叫重疊克隆群)。
跨疊克隆群的製作方法有三種:
1)菌落雜交法。其基本原理是利用基因組某一區域特有的或與所需分離的目標基因緊密連鎖的DNA分子標記為探針篩選大片段基因組文庫,可以分別得到一系列的陽性克隆,這些克隆之間有部分片段是重疊的,根據其重疊部分可以把它們有序地呈線狀排列成跨疊群(contig)。在兩個跨疊群之間會出現空隙,需要通過染色體步行來填補。該技術從分離大片段陽性克隆群的左、右末端人手,以它們為探針再次篩選基因組文庫,獲得新的克隆即為沿著染色體向前走了一步,重複這一過程,就能一步一步地將空隙填滿,將兩個克隆群整合在一起,這一方法能利用各種分子標記掃描文庫,準確得到攜帶分子標記的陽性克隆,是構建跨疊克隆群的首選策略。
2)PCR法。即以PCR技術為基礎發展了一系列技術,包括STS、RAPD、AFLP和Alu-PCR等已被廣泛應用於跨疊克隆群的構建。STS策略是利用已知核酸序列,從兩端合成兩個與其配對的引物,利用PCR進行擴增,理論上講具有相同擴增帶型的克隆,也可以說共享一個或數個STS位標的克隆肯定是跨疊的。這個方法具有簡便、快速、精確度高等優點,是人類基因組計劃所採用的主要策略之一。另一重要的基於PCR的策略是A1u-PCR技術,該技術利用人類基因組的Alu重複序列,合成一對特異性引物,進行PCR擴增,然後以PCR產物為探針,從文庫中篩選出陽性克隆。這一方法簡便,但有時會出現一些誤差,當與限制性內切酶物理圖譜結合使用時,就可以比較精確地排列出陽性克隆的重疊順序。
3)DNA指紋—錨標法。其原理是首先對隨機克隆的DNA選定一個6鹼基識別位點的限制性內切酶消化,然後用放射性同位素進行末端標記,經標記后DNA片段用另一個4個鹼基識別位點的限制性內切酶消化,用序列分析膠分離這些片段,然後經放射性自顯影檢測。這些指紋圖譜數據經計算機處理后,根據片段的相似性,構建跨疊克隆群。
熒光原位雜交技術(Fiber-FISH)是最近幾年新發展的方法,它使FISH技術的分辨力接近其理論值1Kb,在光學顯微鏡的識別範圍內。Fiber-FISH適宜於基因組的數量作圖,而且由於可以在熒光顯微鏡下同時觀察幾種探針的位置和順序,將有效地排除染色體步行過程中經常遇到的復重序列帶來的因難。這些物理圖譜的構建,無疑給圖位克隆感興趣的基因奠定了堅實的基礎。
(3)構建高質量的基因組文庫
在基因的圖位克隆技術中,高質量的基因組文庫的構建也是克隆成功的另一關鍵。圖位克隆的基因組文庫主要有Cos質粒文庫和人工染色體(YAC)文庫。作為被克隆的載體。可以採用Cos質粒,它是1種含有λ噬菌體的cos位點的質粒載體,大小在10kb左右,可高效地克隆25~35kb的DNA片段。當要克隆大片段DNA時。需要YAC作載體,以YAC為載體,可將300~1000kb的DNA片段克隆:因此,以YAC為載體可以構建高等植物的完整基因組文庫。除YAC外。後來又陸續發展了BAC、PAC等幾種以細菌為寄主的載體系統。
圖位克隆在理論上適用於任何物種,但目前主要還是用於真核生物。真核生物一般都有內含子,一個基因往往長達幾個到幾十個kb,只有容納大片段插入子克隆載體,才能攜帶完整的基因。柯斯質粒從λ噬菌體改造而來,由於採用質粒的複製子,柯斯質粒克服了包裝的限制,插人片段可達40~50kb。雖然如此,柯斯質粒還是不敷應用,就又發展了人工染色體技術。人工染色體在細胞周期中可以正常複製,配對和分離。作為載體,人工染色體的插入片段可達2Mb左右,能夠覆蓋完整的真核基因。最早發展的人工染色體是酵母人工染色體(YAC)。1983年,Murray等首次構建了總長度為55kb的YAC。1987年,Burke等得到了能夠克隆大片段DNA的YAC載體,證明YAC可以構建高等生物的完整基因組文庫。YAC具有比較大的容納能力,但不太穩定,嵌合體比例也較高,而且難以與酵母自身的染色體分開。後來陸續發展了P1、BAC、PAC等幾種以細菌為寄主的載體系統。值得一提的是進展一直緩慢的哺乳動物人工染色體(MAC)和人類人工染色體(HAC)也已取得突破性進展。
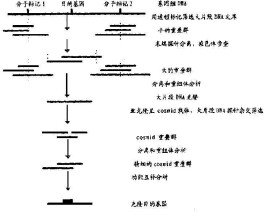
1.6染色體步行、登陸
找到與目標基因緊密連鎖的分子標記(最好分佈在基因兩側)和鑒定出分子標記所在的大片段克隆以後,接著是以該克隆為起點進行染色體步行,逐漸靠近目標基因,以該克隆的末端作為探針篩選基因組文庫,鑒定和分離出鄰近的基因組片段的克隆,再將這個克隆的遠端末端作為探針重新篩選基因組文庫。繼續這一過程,直到獲得具有目標基因兩側分子標記的大片段克隆或重疊群。如果目標基因所在區域已經完成分子作圖,就有一套現成的順序排列的大片段克隆可以利用。當遺傳連鎖圖譜指出基因所在的特定區域時,即可取回需要的克隆,獲得目標基因。
染色體步行的主要困難在於,當必須經過一個無法克隆的片段時(如對寄主無害),步行的過程會被打斷;當克隆的一端是重複序列時,步行的方向會步入歧途。為了克服這些困難,發展了染色體登陸、跳步和連接等方法。染色體登陸是找出與目標基因的物理距離小於基因組文庫插入片段的平均距離還小的分子標記,通過篩選文庫直接獲得含有目標基因的克隆,完全避開染色體步行的過程。染色體跳步—連接是使用一個識別位點很少的隨機一個識別位點很多的酶構建跳步文庫和連接文庫。跳步文庫的插入片段是大片段克隆末端經過雙酶切的部分,由同樣的文庫進行克隆。連接文庫的插入片段是由切點較少的酶產生的,具有切點較少的那個酶的識別位點。在染色體步行的過程中,交替應用兩個文庫進行跳步和連接,最終逼近目標基因。
1.7鑒定目標基因
篩選和鑒定目的基因是圖位克隆技術的最後環節,找到與目的基因緊密連鎖的分子標記並鑒定出分子標記所在的大片段克隆后,接著是以該克隆為起點進行染色體步行,逐漸靠近目的基因。當遺傳連鎖圖譜指出基因所在的特定區域時,即可取回需要的克隆,獲得目的基因。在與目的基因緊密連鎖的分子標記及大插入片段基因組文庫都具備的情況下,就可以以該分子標記為探針通過菌落雜交,藍、白斑挑選的方式篩選基因組文庫而獲得可能含有因的基因的陽性克隆。在用覆蓋目的基因區域的大片段基因組DNA克隆篩選區域特異的cDNA后,仍需對目的基因編碼的cDNA作進一步證實。現有證實目的基因編碼的cDNA克隆的方法有:1)精細作圖來證實某候選基因克隆與目標性狀共分離;2)證實某候選基因克隆的時空表達模式與目標性狀的表現相同;3)把候選基因序列與現有已知功能基因序列資料庫進行同源性比較,推斷其功能;4)比較候選基因序列在野生型與突變型之間的差異,確定在二者之間變異的cDNA序列;5)轉化候選基因。
這些陽性克隆中可能含有多個候選基因,從一系列候選基因中鑒定基因是定位克隆技術的最後一個關鍵環節。現在最常用的方法是用含有目標基因的大片段克隆如BAC克隆或YAC克隆去篩選cDNA文庫,並查詢生物數據信息庫,待找出候選基因后,把這些候選基因進行下列分析以確定目標基因:1)精確定位法檢查cDNA是否與目標基因共分離;2)檢查cDNA時空表達特點是否與表型一致;3)測定cDNA序列,查詢資料庫,以了解該基因的功能;4)篩選突變體文庫,找出DNA序列上的變化及與功能的關係;5)進行功能互補實驗,通過轉化突變體觀察突變體表型是否恢復正常或發生預期的表型變化。功能互補實驗是最直接、最終鑒定基因的方法。
基本信息
- 中文名
- 圖位克隆法
- 外文名
- Map-based cloning
- 別名
- 定位克隆
- 拼音
- tú wèi kè lóng fǎ
- 出處
- 分子生物學
- 提出者
- Alan Coulson
- 意義
- 最常用的基因克隆技術
- 方法
- 新型分離和克隆植物基因