晶體化學
晶體化學
研究晶體在原子水平上的結構理論,揭示晶體的化學組成、結構和性能三者之間的內在聯繫以及有關原理的物理化學分支學科。
在組成、結構和性能三者中,關鍵是結構這個環節,它上承組成,下啟性能。組成通過結構的中介而聯繫性能,可由晶體化學中相當普遍存在的同分異構現象為例來說明。如方解石和文石的成分同屬碳酸鈣,但因其離子結合方式不同而具有迥然不同的晶系和解理性。金剛石和石墨均由碳元素組成,但前者為透明、硬度極高的絕緣體,可用作地質鑽探用的耐磨材料;而後者為黑色、硬度極低的良導體,可用作電極和“鉛”筆芯的主要材料。兩者幾乎對立的性質,起源於其內部鍵型、構型不同。金剛石中碳原子通過定域的共價鍵連結成架型的結構;具有層狀結構的石墨及其層分子中π電子的離域則與石墨之低硬度和高電導率相聯繫。20世紀50~60年代,曾廣泛應用的殺蟲劑六六六有α、β、γ等異構體(或稱變體),其中只有γ變體才具有藥效。活性蛋白與變性蛋白的同分異構現象也是重要的實例,蛋白活性的喪失起因於高級結構發生的變異。其他例證還有成分同為 1,4-聚異戊二烯的合成橡膠和古塔波膠;廣泛用作催化劑載體的氧化鋁的γ變體與α變體等。
晶體化學原理首先涉及鍵型、構型以及它們隨組成而變異的規律,其原理的表達主要通過組成晶體結構的原子、離子的數量關係、大小關係和作用力的本質及其變異等要素來進行。性能中首要的是決定某一物質或化合物能否存在的穩定性,而晶體及其所包含的分子的其他物理或化學性質也無不由其結構來決定。現代晶體化學是在大量實測系列晶體結構信息的基礎上總結出規律的。因此,它一方面有其堅實的實踐基礎,另一方面能對材料科學、合成化學、生物化學、地球化學和礦物學等相鄰學科起重要的指導作用。
晶體化學起源於晶體學向化學的滲透。在晶體學發展的經典階段,人們還只能從觀察晶體的多面體的外形來聯繫晶體的組成和結構。但這種聯繫也曾對化學的發展作出巨大的貢獻。1819年德意志化學家E.米切利希發現異質同晶現象。在當時,很多元素還只有當量而不知其原子量,這一發現曾起過與杜隆-珀替定律相仿的重要作用。在1850年前後,L.巴斯德注意到了酒石酸鹽晶體的旋光性與其外形中缺乏對稱中心和鏡面這一事實間的聯繫。他在顯微鏡下拆分了手征性不同的兩種酒石酸鹽晶體。這一發現對有機物立體化學的發展有過深刻的影響。在19世紀下半葉,聯繫晶體化學組成與晶體外形及晶面夾角數據的工作,積累不少,主要概括於德國晶體學家 P.H.von格羅特1919年出版的《化學晶體學》與俄國Ε.С.費德羅夫1920年出版的《晶體界》兩書之中。
1912年德國M.von勞厄對晶體X射線衍射效應的重要發現,實為晶體學發展進程中的一個里程碑。它為X射線晶體學的誕生奠定了基礎,從而使經典晶體學過渡到現代晶體學。在X射線晶體學的初創時期,即使像氯化鈉等簡單離子化合物的結構,對於化學家來說還是個難題,他們套用有機結構理論中關於原子價和分子等概念而陷入困境。但在1913年,離子化合物氯化鈉和無機單質金剛石在晶體學家手中卻是作為最簡單的結構問題予以解決了。此時化學家才明白,在這些簡單無機晶體中並不存在分立的分子集團。這些重要而又屬於啟蒙性的晶體結構知識為無機物的晶體化學開闢了良好的前景。基於這一歷史背景,在1913~1929年這一時期,晶體學家選擇無機單質和離子化合物作為主要對象,進行了相當系統的研究。在這個時期中,W.科塞爾、G.N.路易斯、N.V.西奇威克和I.朗繆爾提出和發展了關於電價結合(見離子鍵)和共價結合(見共價鍵)的理論。離子半徑的主要研究者有德國的V.M.戈爾德施米特和美國的L.C.鮑林等。離子化合物點陣能問題的主要研究者有M.玻恩,A.朗德,F.哈伯和E.馬德倫等。離子極化和變形的主要研究者是K.法揚斯。1927年戈爾德施米特在簡單離子化合物晶體結構材料的基礎上,提出了他的晶體化學定律:“晶體的結構取決於其組成者(原子、離子和原子團)的數量關係、大小關係和極化性能。”對於離子化合物來說,定律中所說組成者的數量關係是指正、負離子的數量比,組成者的大小關係是指正、負離子的半徑比,組成者的極化性能主要是指負離子的可極化性和正離子的極化力(負離子電價低,半徑大,一般易被極化;正離子半徑越小,極化能力越大)。當正、負離子間極化因素增強時,離子鍵將在一定程度上向共價鍵過渡,從而導致產生鍵長縮短、鍵能遞增、正離子配位多面體偏離高對稱性、產生畸變等效應。晶體化學定律高度概括了決定化合物結構型式的組成者的三個結構要素。在無機化合物的晶體化學中,一般按化學式(即組成比)的類型AB、等分類進行討論。就同一類化學式的化合物來說,戈爾德施米特將晶體結構型式隨組成者大小關係和極化性能的遞變而產生的變化稱為型變。事實上晶體的結構型式還將受溫度、壓力等外界條件的影響。同一化合物或單質在不同條件下可生成不同型式的同分異構體。這種現象又稱為多晶型現象。戈爾德施米特定律的概念極為清晰,但其適用範圍主要局限於組成比簡單的無機化合物。1913~1929年,以W.L.布喇格和鮑林為代表的晶體學家,從事以硅酸鹽為主體的大量複雜含氧酸鹽的晶體結構研究。這些研究促進了無機晶體化學第一次繁榮的高潮,它以鮑林總結、提出的五個關於離子晶體結構的鮑林規則為標誌。在鮑林規則的表述中,突出了形成離子配位多面體的原理及制約配位多面體間相連接的規律,並將它與離子晶體結構穩定性的問題聯繫起來。在離子晶體中,正離子應當位於負離子形成的配位多面體(正四面體、正八面體、立方體、立方八面體等)的中心。第一規則涉及“負離子配位多面體的大小和型式主要取決於正、負離子半徑和與半徑比”的問題。第二規則即電價規則,是五個規則的核心,它涉及多面體頂角如何公用的問題。規則定義每個離子鍵的靜電鍵強度s為離子電價ω 除以配位數v(即)。對穩定的離子晶體,負離子的電價將接近或等於其鄰接諸離子鍵的鍵強之和。第三規則涉及多面體公用棱和面將降低結構穩定性的問題。第四規則涉及什麼樣的正離子多面體不鄰接的問題。第五規則要求同一種離子的結合方式趨於最少。戈爾德施米特定律和鮑林規則等晶體化學原理對無機化學、礦物學、水泥陶瓷工業等的發展起了重大的推動作用。到了70年代,著名的鮑林電價規則已被以加拿大I.D.布朗為代表的晶體化學家進一步發展為價鍵理論。這一理論對複雜無機化合物的結構化學有重大的指導意義。
按晶體化學的分類系統,無機物的晶體主要劃分為單質、二元化合物、多元化合物、含氫化合物、合金等體系。
對非金屬單質,因其中定域共價鍵佔主導地位,起支配作用的結構化學規律是8-N規則。N是非金屬元素所屬的族數,是指每個原子與鄰近原子可形成共價(單)鍵的數目。如對硫和硒,,則每個硫或硒原子鄰接原子數為,因而硫和硒可形成環狀或鏈狀的分子。
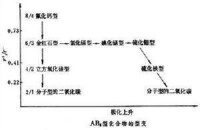
型變規律
簡單二元離子化合物的典型結構有氯化鈉型、氯化銫型、立方硫化鋅型、六方硫化鋅型、氟化鈣型和金紅石型等。在一般場合,因負離子的半徑大,它在佔據空間上起主導作用,因而多採取A1型、A3型或其他的緊密堆積方式,而正離子則按正、負離子半徑比而佔據負離子在密堆積中所形成的四面體、八面體等多面體孔隙。例如,氯化鈉的結構可描述為氯離子作A1型立方最密堆積,而鈉離子 Na+則佔滿全部Cl-所形成的八面體空隙。在氟化鈣中,F-作簡單立方堆積,而所有Ca2+離子則佔據半數由F-所形成的立方體空隙。上述實為兩種半徑不同的圓球密堆積問題。對於二元離子化合物,由於整個晶體必須保持電中性,正、負離子的電價比和配位數比必然受正、負離子數量比的制約如下:關於正、負離子半徑比r+/r-和極化因素變遷對結構型式的影響,可以AB2型化合物的型變規律為例說明之(見圖)。當r+/r-下降時,極化程度將上升而導致高對稱的離子性結構氟化鈣型和金紅石型通過過渡的二氧化硅型而向分子型的二氧化碳結構型轉化。另外,隨著過渡元素極化力之增強及負離子可極化性的上升,高對稱的構型將通過氯化鎘、碘化鎘、硫化鉬等層型結構向島型的結構型過渡。多元化合物的類型甚多,包括各種簡單和複雜的含氧酸鹽、各種金屬配合物和簇合物等。對於離子性成分高的化合物晶體,鮑林規則具有重要的指導作用。對於原子簇金屬化合物,晶體結構所提供的原子鍵合方式和關於鍵長、鍵角的信息將對成鍵本質的了解和成簇規律的總結提供重要的依據。例如,一般可根據金屬原子間的距離來判斷是否有含金屬鍵成分的M-M鍵的存在等。
對於含氫體系,如酸、酸性鹽、氫氧化物、水合物等,需要強調的是最大限度地形成氫鍵的晶體化學原理。
在合金體系中,佔主導地位的組成者是電負性小(或電正性大)的元素。合金中的物相一般可分為金屬固溶體和金屬化合物兩種類型,金屬化合物又分為組成可變和組成確定的兩種。一般,組成者的電化學性質(主要指電負性)、原子半徑、單質結構型式越相近,則生成固溶體的傾向越大;電化學性質和原子半徑相差越大,則生成金屬化合物的傾向就越大。合金體系一般多用多晶粉末法結合相圖、化學圖進行研究。合金體系的晶體化學與材料科學關係密切,在國民經濟中有重要意義。
蛋白質晶體化學,研究蛋白質晶體結構的物理化學分支學科。蛋白質分子是由上百或更多的α-氨基酸作為單體縮合而成的多肽(見肽)鏈構成的。能構成蛋白質中多肽鏈的α-氨基酸總共有 20種L-氨基酸。通過它們不同的組合和排列形成氨基酸順序不同的多肽鏈,然後這些多肽鏈進一步通過交聯構成千萬種蛋白質分子。
晶體化學的發展與有機化學關聯的密切程度並不亞於無機化學。在它發展的前期,涉及有機化合物的代表性研究工作有1923年R.G.迪金森測定第一個有機物晶體六亞甲基四胺的結構;1947年C.W.布恩對耐綸66晶體結構的研究;1949年D.克勞富特等完成了青霉素衍生物苄青霉素的結構研究;1952年初步測定了第一個夾心式金屬有機化合物二茂鐵的晶體結構;在40~50年代,蘇聯晶體化學家Α.И.基泰戈羅茨基在有機物的晶體化學上也取得很大的成就,1955年他曾出版了《有機晶體化學》一書。
自1966年以後,由於計算機控制的自動單晶衍射儀和與之匹配的晶體結構分析軟體的迅速發展和普及,X射線晶體學方法成為取得有機分子立體結構和鍵參數最有效和得力的工具。1977年所測有機化合物和金屬有機化合物的晶體結構數量已超過三千項。這些大量的晶體化學信息已為深入研究有機反應機理、指導合成和深入探討有機物分子構型和構象與分子化學活性間的內在聯繫提供了可靠的依據。
晶體化學家在生物大分子結構研究中的貢獻也是巨大的。鮑林在1951年提出了多肽的 α螺旋體。J.D.沃森等受此啟示,進一步在1953年提出脫氧核糖核酸雙螺旋模型,初步解開了遺傳信息之謎。1957年J.C.肯德魯發表了具 6埃解析度的肌紅蛋白的結構。這使人們第一次看到一個蛋白分子的立體圖像。1959年M.F.佩魯茲經過多年的奮鬥終於用同晶置換法解出了血紅蛋白的結構。這兩大發現,為肌紅和血紅蛋白的載氧功能的闡明提供了結構基礎。1965年D.菲利普斯測定了溶菌酶的三維結構。1967年W.N.Jr.利普斯科姆測定了羧肽酶 A的結構,揭示了酶功能專一性問題。到1986年為止,用晶體學方法測定生物大分子的結構累計已達280個左右。
晶體化學在近代自然科學中的地位可簡單地歸納如下:①晶體化學起源於晶體學向化學的滲透;②因很多材料(如合金、分子篩等)只存在於晶態之中,再者,分子立體結構知識的主要來源是晶體結構,所以,當今晶體化學已成為結構化學信息的主要源泉;③晶體化學在當今自然科學中有廣泛的橫向聯繫,它不僅是研究化學反應機理和化合物構效關係的指南,而且已成為材料科學和分子生物學深入發展的支柱。
