CCR5
HIV-1入侵機體的輔助受體之一
作為G蛋白偶聯因子超家族(GPCR)成員的細胞膜蛋白,是HIV-1入侵機體細胞的主要輔助受體之一。
趨化因子CCR5,以CCR5為靶點的HIV-1受體拮抗劑越來越受關注,主要有趨化因子衍生物、非肽類小分子化合物、單克隆抗體、肽類化合物等4類。這些抗病毒活性強、親和力高的CCR5拮抗劑,已有一部分進入了臨床試驗階段。
自1981年發現第一例由人類免疫缺陷病毒(HIV)引起的傳染性疾病———獲得性免疫缺陷綜合症(簡稱艾滋病,AIDS)以來,儘管對艾滋病的臨床治療已有了很大進展,但是仍無有效治癒手段可以攻破此科學難題。研究表明,HIV可分為HIV-1和HIV-2兩種亞型,HIV-1致病力強,是引起AIDS的主要病原體。
目前已有30多種抗HIV-1藥物得到美國食品與藥品監督管理局(FDA)批准,其中17種是逆轉錄酶抑製劑(包括13種核苷類逆轉錄酶抑製劑及4種非核苷類逆轉錄酶抑製劑),11種蛋白酶抑製劑,1種CCR5受體抑製劑(maraviroc),1種整合酶抑製劑(raltegravir)以及1種融合抑製劑(T20)。然而,已被批准的藥物中沒有一種是可以完全抑制病毒感染的,而且由於HIV-1突變株的產生,大部分均對不同類型的拮抗劑具有耐藥性。此外,隨著對病毒入侵過程的深入了解,研究者發現,除了病毒入侵所必須的CD4受體外,重要的輔助受體如CCR5或CXCR4,在gp120與CD4識別後發生的構象變化中起到了至關重要的作用。因此,研究者們漸漸將目光轉移到了這個新的靶點,目前已有幾種CCR5抑製劑正處於臨床前和臨床試驗中,並且還有一套評價利用CCR5拮抗劑來控制HIV-1病毒感染的臨床前試驗方法用於藥物的研究。
病毒入侵過程是一個級聯的結合與構象變化反應,因此,根據病毒入侵複製裂解的不同階段,拮抗劑可分為病毒入侵拮抗劑(如CD4拮抗劑、輔助受體拮抗劑)、逆轉錄酶拮抗劑、融合拮抗劑、整合酶拮抗劑、蛋白酶抑製劑等。而針對輔助受體CCR5的拮抗劑又可分為趨化因子衍生物、非肽類小分子化合物、單克隆抗體、肽類化合物等4類。
病毒特異的CD4+T細胞是針對HIV免疫應答的重要組成部分,也是HIV-1感染過程的首要靶點。自1984年發現HIV-1通過與CD4結合感染宿主細胞,隨後研究又發現僅有CD4分子並不能介導HIV-1的侵入,同時還需要一種或幾種輔助受體。1996年證實,趨化因子受體CXCR4和CCR5是HIV-1感染的輔助受體(coreceptor)。而在2008年2月,美國國立衛生研究院又發現了另外一種新的HIV-1受體———整合素α4β7(integrinα4β7),此受體的發現也許可解釋為什麼消化道為HIV複製的主要場所,同時為艾滋病新葯的開發指明方向。
趨化因子是一類具有趨化活性的小分子細胞因子,它的功能在於使細胞表面具有跨膜G蛋白偶聯受體的細胞聚集在一起,參與體內的免疫平衡作用。迄今為止,已發現人類趨化因子家族有50多個成員,根據其N?末端兩個半胱氨酸殘基的相對位置可分為4個不同結構的家族:CXC(或α)、CC(或β)、C(或γ)和CXC3C(或δ)。儘管CCS和CXCS具有相似的一級、二級和三級結構,但是由於兩者四級結構的差異,它們分別作用於不同的受體。趨化因子受體是屬於鳥嘌呤核苷酸結合蛋白(G蛋白)偶聯的7次跨膜蛋白受體家族,根據其配體可分為CXC和CC受體。迄今已鑒定出28種趨化因子受體,其中包括6種CXCRs(CXCR1~CXCR6)、10種CCRs(CCR1~CCR10)、1種XCR、1種CX3CR。其中的趨化因子受體CXCR4與CCR5,由於它們與HIV-1的侵染有關而備受關注。幾乎所有的HIV-1病毒株都是利用其中的CCR5或CXCR4受體,或同時利用兩種輔助受體侵入細胞的。有研究表明,CCR5和CXCR4的N-末端和第二胞外區對於它們與HIV-1gp120的結合起著關鍵的作用。
利用何種趨化因子受體侵入細胞決定了HIV-1病毒株的嗜性。單核細胞/巨噬細胞嗜性的病毒株利用趨化因子受體CCR5侵入細胞,稱為R5嗜性;T細胞嗜性的病毒株利用趨化因子受體CXCR4侵入細胞,稱為X4嗜性;而既能利用CCR5,又能利用CXCR4侵入細胞的病毒株稱為R5/X4嗜性。HIV-1gp120與何種輔助受體結合是隨著HIV-1侵入靶細胞的不同階段而有所變化的,通常在感染的初期是以CCR5為輔助受體;隨著感染程度的加深,HIV-1由R5嗜性轉化為R5/X4嗜性,而雙嗜性的HIV-1是以CXCR4為主要輔助受體的。Cocchi等人研究預測,病毒包膜蛋白的V3環與HIV-1病毒株的嗜性轉化有關,尤其是V3環中11位及25位的鹼性氨基酸在不同類型病毒株中的差異可以說明一些問題。
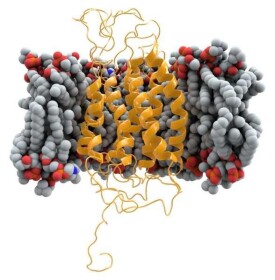
CCR5是細胞內β趨化因子(RANTES、MIP1α和MIP1β)的受體,具有調控T細胞和單核細胞/巨噬細胞系的遷移、增殖與免疫的功能,主要表達於記憶性的靜止期T淋巴細胞、單核細胞、未成熟的樹突狀細胞等的細胞膜上。它的分子量為40.6kDa,由352個氨基酸殘基組成,結構上可分為:胞外N末端,3個胞外環(EL13),3個胞內環(IL13),7個跨膜α螺旋和胞內C末端(圖1)。CCR5除了作為HIV1早期侵染所必須的輔助受體外,有研究顯示,CCR5還對增強長期同種異體移植存活率發揮重要作用,CCR5拮抗劑還可以有效治療一些臨床疾病,包括器官移植、哮喘、風濕性關節炎、多發性硬化症與糖尿病等。
從病理角度看,CCR5輔助HIV-1感染的過程是一個多區域參與的複雜過程,gp120和CCR5的相互作用可能遵循兩步結合機制:第1步,CCR5的N末端區域採取恰當的構象識別並結合gp120;第2步,gp120與CCR5的構象發生變化,CCR5的第二胞外區與gp120的V3區相互作用,最終導致膜融合和病毒遺傳物質進入細胞。CCR5作為HIV-1的重要輔助受體參與膜融合過程,其N末端和EL2主要與gp120相互作用,而N末端對於HIV-1的感染來說是必需的。CCR5與趨化因子或gp120的結合位點是相互獨立的,或者有部分重疊,產生的不同功能主要是通過極性、疏水性氨基酸的三維構象的細微調整來實現。此外,gp120、趨化因子和各類CCR5拮抗劑在CCR5上的結合位點也並不完全相同。
從基因角度看,在CCR5基因中存在的多個突變體能夠有效抵抗HIV的病毒感染以及延緩病情的惡化。目前已在此基因的編碼區和啟動子區域發現了多種有意義的突變,尤其以對基因編碼區突變體CCR5△32的研究最為廣泛,CCR5△32是指CCR5基因的32個鹼基缺失的突變,即在CCR5等位基因編碼區域第185位氨基酸密碼子以後發生了32個鹼基缺失,導致讀碼框架錯位,缺失了與G蛋白信號通路相關的胞外第三環結構,從而使CCR5蛋白無法正常穿膜表達於細胞膜上,進而使HIV1的gp120不能與CCR5△32有效結合,使HIV1病毒不能進入宿主細胞。人群調查和實驗研究結果表明,CCR5△32缺失的個體擁有正常的免疫功能和炎症反應,並且對HIV-1的感染表現出顯著的抵禦能力,因此,作用於CCR5的抑製劑將有效阻斷HIV?1感染。
從氨基酸組成上看,通過點突變方法並聯用了病毒侵染試驗來進行基於CCR5同源模型的預測以及分析其中的30個氨基酸殘基的功能,研究者發現一組關鍵的芳香族和脂肪族氨基酸殘基具有與配體結合的疏水性核心。這些發現為解釋拮抗劑與CCR5如何作用提供了一個結構基礎。對於小分子非肽類CCR5拮抗劑而言,CCR5的7個跨膜區域(H1~H7)與小分子拮抗劑的作用過程相關,跨膜結構域中的酸性氨基酸尤其是一些芳香族和脂肪族氨基酸,它們通過H1~H3和H7的螺旋區線性形成一個用於小分子拮抗劑結合的小腔。
CCR5作為HIV?1受體拮抗劑的理想靶點,其拮抗劑藥物抑制R5嗜性的HIV?1感染細胞的機制是,它們與CCR5結合后,使CCR5構象發生變化不利於gp120的識別或者導致CCR5的內源化作用(internalization),阻斷了HIV?1與細胞包膜蛋白結合,導致HIV?1與CCR5在細胞表面結合的數量減少,從而起到抗感染作用。同時,有些學者擔憂靶向於CCR5輔助受體的抗病毒治療是否會加快R5嗜性病毒株向X4嗜性病毒株的轉變。在體外實驗中顯示,除了一種耐葯株產生嗜性轉換外,大部分對CCR5拮抗劑產生耐葯的HIV?1突變株仍然保持原先的輔助受體嗜性。而且臨床上有限的數據積累亦顯示,已開發的CCR5拮抗劑在臨床試驗中還未發現有加快耐葯株嗜性轉換的病例。因此,CCR5拮抗劑的應用是不會加劇病情的惡化反應。目前,主要的CCR5受體拮抗劑有以下幾種。
β趨化因子(RANTES,MIP?1α及MIP?1β)作為CCR5的天然配體是HIV?1受體當然的拮抗劑,在一定程度上可以保護細胞免受HIV?1的感染,其主要作用機制是誘導了CCR5的內吞作用(endocytosis)。然而,由於天然趨化因子具有半衰期短(<10min)以及潛在的炎症應答作用,它們作為拮抗劑藥物並不是很合適。也有報道顯示,高濃度的CC?趨化因子能夠減緩病情的惡化,但是Marozsan等研究人員同時也發現高濃度的CC?趨化因子也可通過活化細胞而增強HIV?1的感染。而且Mosier等人發現CC?趨化因子也可促進R5嗜性病毒株向X4嗜性病毒株的轉換以致病情加劇。因此與天然的配體相比,趨化因子衍生物通過不誘導信號通路而使受體內化的方式阻斷相應的受體表位而更具優越性。
RANTES(3?68)是缺失了RANTES的N?末端2個氨基酸殘基的CCR5拮抗劑,具有抑制R5嗜性HIV?1侵入的活性;RANTES(9?68)是缺失了RANTES的N?末端8個氨基酸殘基的CCR5拮抗劑,但是相比於RANTES(3?68)的抑制活性較低;AOP(氨基氧戊烷)?RANTES是通過將AOP基團偶聯到RANTES的氨基末端得到的一個很強的CCR5拮抗劑,在AOP?RANTES的作用下,CCR5被修飾后內吞,細胞表面的表達量不可逆地減少,因此具有抑制R5病毒株感染的作用,並且沒有誘導趨化的功能。此外,以RANTES為基礎的修飾衍生物還有(NNY)?RANTES,Met?RANTES,PSC?RANTES。其中以PSC?RANTES的特異性阻斷R5HIV?1的作用最強,其抗病毒能力是AOP?RANTES的50倍左右。Lederman等利用嵌合性猿/人免疫缺陷病毒SHIVSF162(R5嗜性)進行研究的結果證實,PSC?RANTES能有效地防止HIV通過陰道途徑傳播。幾乎所有的RANTES修飾衍生物均具有使CCR5受體內源化並下調細胞表面的CCR5表達量的作用,從而達到潛在的抗病毒活性。LD78β(CCL3L1)是MIP?1α的同種型,兩者的區別是僅有3個氨基酸不同,但是誘導細胞內Ca2+釋放及增強細胞趨化作用的能力遠強於MIP?1α和RANTES,它能特異性地與CCR5結合,可以下調CCR5在人類單核細胞和巨噬細胞上的表達,抑制R5病毒株的感染,編碼CCL3L1的基因拷貝數的多少影響細胞對HIV的易感性,拷貝數越少,細胞的病毒感染率越高;而此後研發的AOP?LD78β具有比AOP?RANTES高10倍的抑制病毒入侵活性,因此可能是最有效的趨化因子衍生物。2002年又發現,HCC?1(人類CC趨化因子1)的蛋白水解產物HCC?1作用於CCR5的EL2,它可以促進T淋巴母細胞、單核細胞、嗜酸性粒細胞的趨化作用和Ca2+釋放,同時也能介導CCR5內吞作用。熒光細胞活性實驗顯示,HCC?1能使CCR5在細胞表面的表達量下降75%,並且高濃度的HCC?1具有很強的抗HIV?1的活性。vMIP?I,vMIP?II及vMIP?III是由人皰疹病毒8(humanherpesvirus8,HHV?8)編碼的病毒性趨化因子,它們與巨噬炎症因子(MIP?1α)有25%~40%個氨基酸的相似性,與炎症免疫應答相關。Boshoff等人於1997年在Science發表研究顯示,vMIP?I和vMIP?II均具有拮抗HIV?1入侵有關的輔助受體的作用。其中,vMIP?I主要作為CCR8的顯效劑,但Willey等人研究發現vMIP?I也可在輔助受體缺失或被封閉的情況下抑制HIV?1病毒的入侵;vMIP?II可以與CC和CXC趨化因子受體作用並抑制由CCR1,CCR2,CCR5,CCR8,CXCR4和XCR1介導的HIV?1入侵,可阻斷正常血流狀態下人微靜脈內皮固定的RANTES誘導的單核細胞和Th1的牢固粘附和跨膜移動;而vMIP?III主要作為XCR1的顯效劑發揮作用。
目前以非肽類小分子化合物CCR5拮抗劑的研究佔據主導地位,這類小分子拮抗劑不具有潛在的炎症應答效應,並且具有比生產小分子蛋白成本低,可通過靜脈注射方式給葯的優勢,但是它也具有不能下調受體表達的缺點。非肽類小分子化合物CCR5拮抗劑的抑制效果還與細胞的類型和細胞表面的CCR5濃度相關。非肽類小分子化合物拮抗劑主要有TAK?779,TAK?220,TAK?652,SCH?351125(SCH?C),SCH?417690(SCH?D),GW873140,maraviroc(UK?427875),NIBR?1282和AMD3451等幾種。在小分子CCR5拮抗劑的研究中,許多生物製藥公司參與了進來。TAK?779是日本Takeda公司研發的第一種CCR5小分子拮抗劑,主要用於以CCR5為靶點的抗HIV藥物研究。它屬於季銨衍生物,可以阻斷膜融合階段gp120與CCR5的結合,其作用位點在受體的細胞外側面,跨膜α螺旋1,2,3,7形成的袋狀結構內,這說明CCR5的跨膜結構域同樣是拮抗劑可以選擇的位點。臨床研究表明,TAK?779有很強的CCR5拮抗作用,可阻止CCR5介導的鈣離子信號傳導;它還能抑制利用CCR5進行膜融合的R5型HIV?1在外周血單個核細胞(PBMC)中的增殖,從而起到抗HIV?1的作用。但是由於它的可變抗病毒活性和低效的口服生物有效性,它並不是一種好的抗HIV?1藥物。此外,基於TAK?779的其它兩種小分子拮抗劑有TAK?220和TAK?652,它們均具有較強的生物學活性,其中TAK?652已進入臨床Ⅰ期試驗。而吡咯烷類化合物TAK?220,它能與CCR5上的4,5,6跨膜區結合,只抑制RANTES和MIP?1α與CCR5結合,不抑制MIP?1β與CCR5作用,選擇性高,在小鼠和猴中的口服利用度分別為9.5%和28.5%,而且葯代動力學性質很好,在小鼠淋巴液中的濃度是在其血漿中的2倍,現也已進入Ⅰ期臨床研究。SCH?351125(SCH?C)是小分子肟哌啶類化合物,是高特異性的CCR5拮抗劑,對CCR5有很高的親和性,它與CCR5的1,2,3,7跨膜區結合從而改變CCR5胞外區的構象,本身並不誘導Ca2+釋放,但可以有效地拮抗RANTES的誘導作用,嚙齒類和靈長類動物的體內實驗表明,SCH?351125的口服生物利用度為50%~60%,血漿半衰期為5~6h,R5病毒株的複製顯著減少,它是第一個進入臨床的小分子CCR5拮抗劑。SCH?351125進入Ⅰ期臨床試驗后,發現在高濃度時有延長心臟QT間期的副作用,因此對SCH?351125進行結構改造后得到一系列化合物,如基於SCH?351125的衍生物小分子拮抗劑SCH?417690(SCH?D),它們具有比SCH?351125高10倍的拮抗活性。目前由Schering Plough公司開發的SCH?417690已進入了臨床Ⅲ期試驗。體外活性實驗證明,SCH?417690具有比SCH?351125更強的活性(大約10倍的生物活性),且無心血管反應。SCH?417690具有很好的藥物動力學特性,100%的生物藥效率,84%的藥物結合率,而且不會引起對肝臟酶類的抑制反應。SCH?417690是趨化因子RANTES的一種拮抗劑,當二者同時存在時會相互干擾對方的結合活性。
AMD3451屬於CCR5/CXCR4的拮抗劑,是第一個對CCR5與CXCR4受體都進行拮抗的小分子抑製劑。研究顯示,AMD3451對R5嗜性、X4嗜性及R5/X4嗜性的細胞均具有較強的抗病毒活性。它能抑制CXCR4受體的配體因子SDF?1和CCR5的配體因子RANTES引發的胞內Ca2+信號傳導作用。NIBR?1282是一種CCR5的新型拮抗劑,動物實驗顯示,此拮抗劑具有良好的口服活性,並且在10mmol/L的高劑量下對心臟也不會產生不利的的影響。
GW873140作為一種新的螺環二酮哌啶類小分子拮抗劑,是由GSK公司開發的。此拮抗劑能有效阻斷gp120/CCR5複合體的形成,以及具有對HIV?1很強的抑制活性。藥物代謝動力學實驗顯示,在嚙齒類動物中,GW873140具有較好的口服有效性。但是2005年Nichols等發現GW873140具有嚴重的肝毒性反應而停止了其Ⅲ期臨床研究。另一種有效的小分子拮抗劑maraviroc是由Pfizer公司開發的萘啶醯胺類化合物,本品可口服,吸收快,通常在口服后0.5~4h內達到最高血葯濃度,目前是唯一被FDA批准上市的CCR5拮抗劑。
在 對小分子非肽類CCR5拮抗劑的研究開發過程中,研究者們發現一些基團的有效添加或修飾將會極大地增強藥物抑制活性及吸收率。如Masaki發現,在含有2?吡啶基團的硫氧化合物中,其間的咪唑基團對於增強抑制活性十分重要。並且經過進一步的化學修飾后,如以咪唑基團或1,2,4?三咪唑基團代替吡啶基團將會增強拮抗劑的結合活性。而像1999年誕生的第一支非肽類小分子CCR5拮抗劑TAK?779,它具有低的口服吸收率的缺點,這是由於部分四價的銨根離子的結構只適於皮下注射,但是改進三價銨鹽衍生物的化學修飾方式以代替四價銨鹽將極大提高口服的效率。此外,由於此藥物中銨鹽的組成可能是導致嚴重副作用的主要原因,科學家通過將無銨鹽結構組成的TAK?779和金納米粒子結合起來的方法進行了有關的實驗,從而消除銨鹽的負影響,實驗證實這一結合不僅加大了此藥物阻止HIV病毒感染實驗室培育的白血球的能力,還消除了此藥物的副作用。Shankaran也發現,小分子雜環上的合適替代基團和乙酸側鏈上的烷基取代基將增加小分子拮抗劑的抗病毒活力。
Shinichi也發現,小分子化合物中的極性基團是拮抗劑與輔助受體高效結合所必需的。在對小分子拮抗劑結合模式的研究中發現,不同的拮抗劑與CCR5的結合部位並不相同。而且,不同HIV?1病毒株對不同小分子拮抗劑的敏感性也不一樣。結合於CCR5跨膜區的不同小分子化合物將非競爭性地阻斷gp120與CCR5的結合。小分子抑製劑通過插入CCR5跨膜區並與之結合而影響了CCR5的H3和H6跨膜螺旋區域的位移,而此螺旋區域恰是CCR5活性所必須的部位。這就勢必會影響CC?趨化因子的結合作用,對此現象的一種解釋是拮抗劑深入與跨膜區域的結合可能阻斷了趨化因子的N?末端與CCR5的作用,另一個解釋是拮抗劑與CCR5的深入結合誘導了CCR5的構象變化而不利於趨化因子的結合。因此,為了保留CCR5天然趨化因子的生理活性,設計的小分子拮抗劑應該避免深入插進CCR5的跨膜螺旋束而導致CCR5活性的喪失。
CCR5存在多種構象及可變性,從構象上的多種變化上看,CCR5的膜外具有一個N?末端及三個胞外環,它可以為單克隆抗體提供多個抗原表位的識別位點,因此CCR5mAbs可以識別CCR5上的多種不同的結合表位。研究表明,CCR5的單克隆抗體與不同的受體表位結合且能產生不同的抑制效應。儘管已有多種CCR5mAbs被報道,已知的單克隆抗體類的CCR5拮抗劑主要有PRO140,mAb004,2D7,3A9,ROAb12,ROAb13,ROAb14,ROAb18和CCR?02等,但是具有有效的抗病毒活性的單克隆抗體卻相當少。其中僅PRO140與mAb004具備較強的抗病毒活性,目前正處於臨床Ⅰ期試驗階段。而2D7正處於前期臨床開發階段。CCR5mAbs作為拮抗劑,它具有人體對藥物的不適應性,潛在的過敏反應,不能使輔助受體內源化,易產生耐葯株及中和抗?抗體的缺點,但是它也具有高靶向專一性,較長的胞質半衰期,利用了不同的代謝途徑,不會幹擾小分子拮抗劑的結合等優勢。幾乎所有的小分子拮抗劑均顯示具有抑制CCR5天然配體結合及發揮功能的作用,而CCR5mAbs具有不同的抗病毒和抗趨化的活性,因此可以協同小分子拮抗劑發揮抗病毒的活性。
PRO140,是鼠源的單克隆抗體,覆蓋於CCR5的多個胞外結構域,在不影響CCR5趨化因子受體功能的情況下,具有封閉CCR5的功能,能夠有效抑制HIV?1的粘附。PRO140有諸多優點,除能與CCR5進行多價結合,在血漿中半衰期較長以外,在實驗中還發現,HIV?1在PRO140的抑制作用下,很少產生抗性病毒株,使PRO140成為非常有治療前景的藥物。單克隆抗體2D7,可與CCR5的EL?2結構域(EL?2A:Lys?171/Glu?172)特異性結合,並且高效識別細胞表面的CCR5分子,能夠最大化地中和HIV,從而起到抗R5嗜性病毒株感染的作用;CCR5?02則是CCR5N?末端結構域特異性的單克隆抗體,它不干擾天然趨化因子如RANTES的結合及其正常生理功能,不下調細胞表面CCR5的表達,不干擾gp120?CCR5的結合作用,而是使CCR5形成二聚體導致構象的變化來阻斷HIV?1的感染。單克隆抗體與CCR5的結合部位同小分子拮抗劑並不相同,單克隆抗體主要識別CCR5EL?2表位,而且它主要在阻斷gp120結合的後續步驟中發揮作用,如CCR5的構象變化以及CCR5的寡聚化過程,具有與小分子拮抗劑不同的阻斷機制。
CCR5肽類拮抗劑與單克隆抗體一樣可與輔助受體CCR5的胞外環結構域識別,但又不同於小分子拮抗劑產生的毒性作用,肽類拮抗劑能夠特異性地與CCR5的特定胞外結構結合而產生抑制作用,不會產生毒性作用,安全性好,但它也具有不穩定易被消化降解的缺點。目前,噬菌體肽庫的篩選技術是肽類化合物篩選的重要方法,同時這種肽類化合物也是拮抗劑研究的一個理想方向。通過噬菌體肽庫的篩選技術發現的與蛋白靶點具有高親和力和特異性結合的肽,具有製造費用較低、活性較高(15~60倍)、穩定性較高、與免疫系統發生不相關相互作用的機會較少、器官和腫瘤穿透性較好等優點,甚至可以和抗體相媲美,成為生物製藥中一種可靠替代品。
2003年上市的T20是一種衍生於HIV病毒的跨膜蛋白gp140的643~678號氨基酸的含有36個氨基酸的小肽,是一種融合抑製劑,它具有抑制gp41糖蛋白的構象變化從而阻斷膜融合的作用。而早於輔助受體的發現,T?肽(DAPTA)已被證明具有無毒性的抗HIV?1的活性。T?肽是gp120V2(185~192位氨基酸)的衍生物,它對R5嗜性HIV?1病毒株的抑制機制是通過與HIV包膜蛋白競爭性結合CCR5來阻斷了gp120/CD4與CCR5的相互作用。此外還有一些衍生於CCR5本身結構的多肽也具有抑制gp120與CCR5的作用。S?肽是含有CCR5的N?末端22個氨基酸的多肽化合物,其中第10位和第14位的酪氨酸經硫酸化修飾,另外第20位的Cys也被換成了Ser。研究提示,S?肽的結合位點可能位於gp120表面的CCR5結合區域,它通過干擾gp120與CCR5的結合而阻斷HIV?1的侵入。環十二肽cDDR?MAP是衍生於CCR5第二胞外環的Arg168~Thr177的小分子環肽,它在N?末端及C?末端分別加入Asp,Gly,並使之連接成環,模擬了CCR5的EL2的關鍵區域的十二肽的構象,從而誘導構象特異性抗體的產生,通過抗體和細胞表面表達的CCR5相互作用以及cDDR?MAP本身與HIV?1的gp120直接相互作用聯合抑制R5嗜性病毒株的感染。王芳宇等採用噬菌體展示技術,從噬菌體隨機12肽庫篩選到與CCR5 特異結合的親和短肽(AFDWTFVPSLIL)。並初步證明該序列與CCR5的第二外環(ECL2)具有特異性結合作用,同時該短肽對RANTES以及抗CCR5單克隆抗體同時具有競爭結合和封閉gp120的結合作用。
輔助受體CCR5是目前抗HIV-1感染的首選靶點,因此對HIV?1利用輔助受體(CCR5和CXCR4)侵入靶細胞機制的逐步闡明將有利於更有效地研製出抗HI藥物。不同的輔助受體在不同類型的細胞中存在不同的構像,因此作為一個抑制趨化因子受體發生作用的試劑,它應該能夠識別與結合該受體的不同結合位點或不同的構象,並且不會影響輔助受體天然的趨化因子配體的正常生理功能。研究已經證實不同類型的CCR5拮抗劑具有不同的阻斷機制。小分子拮抗劑通過改變CCR5EL-2構象使之不能識別HIV?1的V3區達到抑制的效果;而CCR5的單克隆抗體拮抗劑識別CCR5的Nt表位並有效阻斷gp120結合於輔助受體,但不阻斷病毒的入侵;相反,具有識別不同表位的兩種單克隆抗體能高效阻斷病毒入侵卻並不能阻斷gp120的結合。研究者們認為,判斷拮抗劑抗病毒活性的標準不是單一地看拮抗劑與CCR5的結合親和力,其它的一些指標如拮抗劑從CCR5上分離的速率及其它一些生理特性也十分重要。
儘管輔助受體拮抗劑可有效預防HIV-1感染,遏制AIDS的流行,但是該類抑製劑的發展也面臨著挑戰。長期使用一種抑製劑,最終會使HIV-1產生耐藥性,不幸的是,幾乎所有的小分子拮抗劑都會產生耐藥性,也有實驗顯示,耐葯株的產生與HIV-1gp120的200個氨基酸組成有關。通過點突變實驗證明gp120C2?V5結構域(271~386位氨基酸)的關鍵作用,Westby等人發現V3的315~317位氨基酸殘基的缺失與小分子拮抗劑UK?427,857相關。所以,開發有效且防止耐藥性病毒株產生是當前各類拮抗劑需要面對的關鍵問題。由於趨化因子(RANTES,MIP-1α及MIP-1β)受體CCR5與gp120的接觸涉及多個位點(N-末端,EL,TM),因此與其單一位點相互作用的CCR5拮抗劑的發展空間有限;而針對CCR5上多個位點的疫苗及其誘生的抗體將對CCR5起到屏蔽作用,使其失去充當HIV-1輔助受體的能力,並且不會對人體的正常生理功能產生影響,因此針對輔助受體多位點的拮抗劑研究將是大勢所趨。此外,為了研製更具耐藥性的抗HIV藥物,或許將輔助受體抑製劑(尤其是CCR5拮抗劑)與病毒生命周期其它環節的抑製劑如逆轉錄酶抑製劑、蛋白酶抑製劑和融合抑製劑聯合應用,可以最大程度抑制病毒複製,並且聯合用藥是將來治療AIDS的主要方向。
基本信息
- 分子量
- 40.6kDa
- 輔助受體
- HIV-1入侵機體細胞
- 結構功能
- RANTE
- 第一例
- 1981年
- 趨化
- 因子衍生物
- 主要方向
- 治療AIDS