超價分子
超價分子
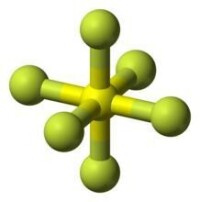
圖中的六氟化硫是一種典型的超價分子
中心原子價電子數為6)、奇電子分子(例如一氧化氮的價電子數是奇數)和超價分子。利用分子軌道理論可以很好地解釋前兩種分子,然而對於超價分子,不但結構沒有得到公認的解釋,甚至定義都處於爭論之中。
超價分子的概念最早由傑里米·穆舍爾(Jeremy I. Musher)在1969年正式提出,他定義以VA族到0族(包括氮族元素、氧族元素、鹵素、稀有氣體)元素為中心原子,而且中心原子氧化態比最低氧化態低的分子為超價分子。
以下是一些較為常見的超價分子的例子:
有機化學中非常有用的高價碘化合物,例如戴斯-馬丁氧化劑(DMP)。
一些四配位(硅除外)、五配位、六配位的硅、磷、硫化合物。例如五氯化磷、五氟化磷、六氟化硫、硫烷以及高價硫烷。
稀有氣體化合物,比如四氟化氙。
一些鹵素互化物,比如五氟化氯。
非經典碳正離子,比如降冰片烷陽離子。
超價分子的N-X-L命名法在1960年提出,經常用於區分超價分子中心原子所在主族,N-X-L的含義分別是:
N為中心原子的價電子數
X為中心原子的元素符號
L為中心原子周圍的配體數目
以下是一些N-X-L命名的例子:
XeF2,10-Xe-2
PCl5,10-P-5
SF6,12-S-6
IF7,14-I-7
關於超價分子本質和分類方法的爭論可追溯到20世紀20年代,即路易斯和蘭米爾時期關於化學鍵本質的爭論。路易斯堅持用普通的二中心二電子鍵(2c-2e)來描述超價分子,從而允許擴大八隅體規則的範圍。但另一方面,蘭米爾堅持八隅體規則,並用離子鍵來解釋超價分子,使得價層電子數仍然為8(比如SF4, F2)。
20世紀20年代晚期及30年代,薩格登提出二中心一電子鍵(2c-1e)的存在性,為超價分子的成鍵提供了無須擴充八隅體規則或引入離子鍵的解釋方法,然而該理論在當時幾乎未被接受。20世紀40年代和50年代時,倫德爾和皮門特爾使三中心四電子鍵理論得到普及,這與薩格登幾十年前的理論本質上是相同的。三中心四電子鍵可被看作兩個共線的二中心一電子鍵組合而成,剩下兩個非鍵電子定域在配體上。
赫爾曼·施陶丁格和格奧爾格·維蒂希在20世紀上半葉進行了製備超價有機分子的嘗試,他們尋求挑戰當時的化合價理論並成功製備了以氮和磷為中心原子的超價分子。超價的理論基礎直到1969年才由穆舍爾基本確立。
1984年,庫策爾尼格總結了前人的文獻,並用大量確鑿的證據證明d軌道參與很少。d軌道參與成鍵最多只有0.3e,而且主要作用是接受配體反饋的電子,增加體系的穩定性。
1990年,馬格努森發表了開創性的成果,明確排除了第2周期元素超價分子中d軌道參與雜化的影響。這是長期以來用分子軌道理論描述這些分子的爭論焦點。這種混亂部分是由描述這些分子的包含d軌道理論基礎造成的(或者說是不合理的高能量以及變形的分子構型),原來認為d函數對分子波函數的貢獻很大。在歷史上,d軌道必須參與成鍵的解釋佔據了統治地位,時至今日,許多教科書上仍然這樣解釋。然而,馬格努森總結自己的工作結果后發現,d軌道的參與與超價基本無關。
里德和施萊爾運用6-31G(d)基組,採用哈特里-福克方程和自然布居分析計算了大量超價化合物的鍵級,表明它們的價層電子數都小於8,符合修正的八隅體規則。
莫利納和杜巴多使用電子局域函數(ELF)研究氟化物,結果說明以氟為配體的化合物中心原子價電子數都小於8。
格萊斯皮和科伯運用多種方法說明超價分子的化學鍵實際上沒有什麼特殊之處,所謂修正的八隅體規則是多此一舉,大量的量子化學計算反而使得人們難以理解它的成鍵。然而,格萊斯皮也發現配體與中心原子電負性相近時,電子基本被均分,實際的價電子數依然超過8,例如Te(CH3)6和Se(CH3)6。
超價分子
缺電子分子通常用兩種方式來使自身穩定。
第一種是形成多中心鍵,使得HOMO電子的離域範圍增大,與LUMO的能級差增加,分子更穩定。例如三氯化鋁經常以雙聚體或多聚體存在,甲硼烷極不穩定易雙聚成乙硼烷。還有碳的配位數超過4時,均形成了多中心鍵,例如中碳的分別配位數為5、6、5、8。
三氟化硼
第二種是將電子反饋給配體。例如三氟化硼中氟的π軌道電子離域到硼的p軌道內,形成通常所說的鍵,此鍵鍵級為1,使得總成鍵電子數為8。而氯化鈹則因為軌道能量相差過大而屬於上一種分子。
對於奇電子分子,通常也有兩種情況。
首先是成鍵軌道或非鍵軌道未填滿,例如氫分子離子H2、二氧化氮可以形成單電子鍵、三電子鍵等等。它們可以得電子、失電子或雙聚形成較穩定的分子。
其次是反鍵軌道上電子不成對,例如常見的氧分子就是一個含2個不成對電子的分子,儘管它的總電子數為偶數。
早期科學家鮑林等人研究超價分子的結構,使用人們熟悉的原子成鍵方式,並運用價層電子對互斥理論解釋了一些問題。因此,AB5和AB6將分別具有三角雙錐和正八面體構型。然而,根據實驗所觀察到的鍵角、鍵長,這種方法明顯違反八隅體規則。此外光譜實驗表明,d軌道能級較高,成鍵釋放的能量不足以補償躍遷消耗的能量。例如,四氟化氙5p和5d的能級差高達10電子伏特,這使得sp3dn雜化根本不可能進行。在這以後有幾個可供選擇的模型被提出。
在20世紀50年代,定域分子軌道理論被引進來解釋超價分子的結構。根據這個理論,五配位、六配位的中心原子將分別發生sp3d雜化和sp3d2雜化,這就要求電子躍遷到空的d軌道上。然而,根據量子化學從頭計算的結果表明,d軌道對超價分子成鍵貢獻其實很小,因而這種模型不太合理,現在也認為這種雜化軌道理論不太重要。這種方法證明,在六配位的六氟化硫中,d軌道基本沒有參與S-F鍵的形成,但是電荷在硫原子與氟原子之間轉移,適當的共振結構可以解釋超價分子。
對八隅體規則的補充已經涉及到超價分子成鍵的離子鍵特徵。作為這些模型的中的一種,三中心四電子鍵模型在1951年被提出,這個模型使用簡單定性的分子軌道來解釋超價分子。3c-4e鍵可以這樣描述,中心原子的p軌道與兩個配體的軌道線性組合形成分子軌道,這導致被佔據的非鍵軌道成為HOMO,空置的反鍵軌道成為LUMO。穆舍爾也推薦這種維持八隅體規則的模型。
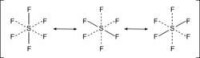
超價分子
對於六配位的分子,例如六氟化硫,六個鍵的鍵長是相等的。合理化的解釋是含有三個3c-4e鍵,每個3c-4e鍵垂直穿過硫氟鍵。
超價分子
這種趨勢也出現在主族元素與含一個或更多孤對電子的配體形成的五配位化合物中,包括以下用氧作為配位原子的五配位硅化合物。
五配位硅化合物中相關的鍵長
五配位硅化合物增大的反應性並沒有得到完整的解釋。科里於和他的同事提出五配位硅原子更高的電正性可能導致了它的反應性增大。初步從頭計算在某些程度上支持這個假設,但也使用了一個小的假設作為基礎。
迪特爾斯和他的同事使用從頭計算的軟體程序Gaussian 86來比較四配位的硅或磷化合物與它們的五配位類似物。這種量子化學從頭計算(英語:Ab initio quantum chemistry methods)方法被用於補充說明五配位化合物的親核反應活潑性增加的原因。對硅來說,使用6-31+G*基組,因為五配位硅化合物是陰離子;對於磷,則使用6-31G*基組。
理論上五配位化合物比類似的四配位化合物親電性更弱,因為配體的空間位阻大和電子云密度高,然而實驗表明它們的親核反應活性比四配位的類似物更強。科學家進行了進一步從頭計算來更深地理解這類四配位和五配位分子的反應現象。不同系列的物質按氟化程度分類。鍵長和電子密度的函數可以表示出中心原子連有氫負離子配體的數目。計算時,每增加一個氫原子就減少一個氟原子。
科學家已經通過這種從頭計算對於四配位和五配位的硅化合物和磷化合物的鍵長、電荷密度、馬利肯鍵重疊進行了計算。四配位硅化合物與氟離子的加合總共增加了0.1個元電荷,這被認為是微不足道的。總的來說,三角雙錐形五配位化合物中的鍵長比類似的四配位化合物更長。Si-F鍵和Si-H鍵的鍵長均有所增加,五配位磷化合物的類似現象則較微弱。硅化合物比磷化合物有更顯著的鍵長增加,這是因為配體有效增加了磷的有效核電荷。
這類化合物是很罕見的,穩定性也較差由於第2周期元素沒有價層d軌道,這使得用雜化軌道理論無法解釋它們的成鍵。但正是它們的不穩定性也說明了d軌道參與成鍵雖少,但也會使體系的能量下降。
硼的五配位化合物也已被合成出來。一個例子中硼與兩個嘧啶環上的電子云密度較高的氮原子形成N-B-N三中心四電子鍵。甚至六配位的化合物也有報道。
主條目:碳鎓離子
甲鎓離子(CH5)等碳鎓離子是五配位碳化合物的典型例子,可以認為其中含有一個三中心二電子鍵。但其中的氫能不斷地發生交換反應,甚至絕對零度下也能發生。這使得甲鎓離子成為一種流變分子,核磁共振氫譜上始終只有一個峰。由於鋰的原子半徑與氫較接近,還存在CLi5離子。
雙分子親核取代反應(SN2)的過渡態是一個超價陰離子。一些特殊的陰離子理論上可以較穩定存在,例如[At—C(CN)3—At]和[Ng—CR3—Ng](Ng代表稀有氣體原子),它們的空間構型都是三角雙錐。
此外,碳與鋰形成的化合物CLi5和CLi6相對穩定,它們分解成CLi4和Li2的過程是吸熱的,其他鹼金屬的類似化合物也有一定穩定性,但不如鋰的這類化合物穩定。
碳的超價化合物還不止這些,例如{[(Ph3PAu)6C]2}、[{Ph3PAu}5C]和一些更加複雜金原子簇化合物。
主條目:五氟化氮和氨
從前,科學家認為五氟化氮分子是不可能存在的,離子型的NF4F也是不穩定的。然而,理論計算表明,三角雙錐型的五氟化氮是三氟化氮與氟分子結合過程中能量最低的狀態。根據分子軌道理論,10個電子分別填充在3個強成鍵軌道(水平方向的鍵)、1個弱成鍵軌道(軸向)、1個非鍵軌道(軸向)中。使用玻恩-哈伯循環計算表明,NF5與NF4F能量僅相差1.0 kJ/mol,都有可能存在。理論上甚至NF6也是穩定的。
此外,氨還能與銨根離子形成超價聚合物,化學式可表示為。這類聚合物可由金屬鉀對[NH4(NH3)n]的單電子還原製得。
主條目:三氟陰離子
由於氟的電負性是除暫無化合物的稀有氣體元素氦、氖外最高的,一般不能形成超價分子。但氟也能在超低溫下形成不太穩定的F3離子,因為F與F2的結合釋放出11 kcal/mol的能量
[編輯]硅
科里於和他的同事對反應特點的早期研究認為四價氯硅烷的水解反應經過了一個超價過渡態。在催化量水存在的情況下測定四價氯硅烷水解反應速率表明,該反應對於氯硅烷是一級反應,而對於水是二級反應。這表明水解過程中兩個水分子與硅烷產生相互作用,因此他們提出了雙親核(binucleophilic)反應機理。科里於和他的同事然後測量了親核催化劑HMPT、DMSO或DMF存在下的水解速率。結果是該反應對於氯硅烷還是一級的,對於催化劑也是一級的,現在對於水也變成了一級反應。水解速率與親核試劑中氧的親核性有關也符合設想。
| 氯硅烷在親核催化劑HMPT、DMSO或DMF存在下的水解速率(溶劑為苯甲醚) | |||||
|---|---|---|---|---|---|
| 氯硅烷 | 親核催化劑 | 水解速率(kobs, Ms,20℃) | 氯硅烷 | 親核催化劑 | 水解速率(kobs, Ms,20℃) |
| Ph3SiCl | HMPT | 1200±100 | MePF2SiCl | HMPT | 2000±200 |
| Ph3SiCl | DMSO | 50±10 | MePF2SiCl | DMSO | 360±50 |
| Ph3SiCl | MDF | 6±1 | MePF2SiCl | DMF | 80±10 |
| Vinyl-1-NpPhSiCl | HMPT | 2200±100 | MePF-1-NpSiCl | HMPT | 3500±400 |
| Vinyl-1-NpPhSiCl | DMSO | 90±10 | MePF-1-NpSiCl | DMSO | 180±20 |
| ( m-CF3Ph)-1-NpHSiCl | DMSO | 1800±300 | MePF-1-NpSiCl | DMF | 40±5 |
| ( m-CF3Ph)-1-NpHSiCl | DMF | 300±50 | |||
將兩者總結后這個小組提出了反應機理:親核試劑(或水)首先進攻四配位硅烷形成五配位的超價過渡態,水繼續進攻該過渡態形成六配位的活性中間體,迅速分解得到羥基硅烷。
霍姆斯和他的同事對硅烷水解進行了進一步研究,四配位的Mes2SiF2(Mes =苄叉苯基)和五配位的Mes2SiF3分別與兩倍物質的量的水反應。二十四小時之後,四配位的硅烷幾乎沒有任何水解跡象,而五配位的硅烷在十五分鐘之內就水解完全了。此外,氟硅烷四乙基銨鹽的X射線衍射數據中二聚硅醇氫鹽的衍射信號支持六配位中間體的存在,此後被迅速取代得到羥基化的產物。該反應以及晶體結構數據支持科里於等人的想法。
硅烷的水解機理以及二聚硅醇氫鹽(左下)的結構
通過格林尼亞反應可知,與類似的四配位化合物相比顯然超價分子的反應性更高。科里於小組通過核磁共振測量了相關的18-冠-6鉀鹽的反應速率,進而獲得了在催化量親核試劑存在下多種四配位和五配位甲基苯基氟硅烷的格林尼亞反應半衰期數據。
| 四配位硅烷以及類似的五配位超價硅烷進行格林尼亞反應的半衰期 | |||||
|---|---|---|---|---|---|
| 氟硅烷 | 親核催化劑 | 半衰期(分鐘) | 氟硅烷 | 親核催化劑 | 半衰期(分鐘) |
| PHMeSiF2 | i-PrMgBr | 780 | PHMeSiF3 | t-BuMgBr | <3 |
| PHMeSiF3 | i-PrMgBr | <5 | PHMeSiF2 | t-BuMgBr | 360 |
| PH3SiF | i-PrMgBr | 320 | PH3SiF2 | i-PrMgBr | 32 |
儘管半反應的方法是不嚴密的,反應速率差別的在誤差範圍內可以繪製出反應的圖解,親核試劑對四配位硅烷的預決速步反應不能完全,始終處於中性四配位物質和五配位陰離子之間的平衡狀態。然後兩分子格氏試劑進行親核配位形成六配位過渡態,產生預計的產物。
四配位硅烷和類似的五配位超價硅烷進行格林尼亞反應的機理
類似的反應性在其他超價化合物中也已經發現,例如多種磷化合物的六配位過渡態已經被證明。正膦和氧代正膦的水解已經得到了研究,說明在水中這是一個二級反應。貝爾斯基等人提出,水的預決速步親核進攻使得五配位磷化合物和六配位的磷化合物處於平衡之中,然後第二個水分子參與的決速步進行質子轉移並開環,得到羥基化的產物。
五配位磷化合物的水解機理
五配位磷化合物的醇解,例如三甲氧基環磷烯在苯甲醇中醇解,也被假定為經過一個類似的八面體過渡態。與水解不同的是,不經過開環這一步。
鹼催化下五配位磷化合物醇解的機理
從這些實驗可以發現,與類似的非超價化合物相比,超價分子的反應活性明顯增加。這可以被歸結於超價分子的結構與同樣超價的過渡態相似,反應過程中所需要的活化能較小。