組蛋白去乙醯化酶抑製劑
組蛋白去乙醯化酶抑製劑
組蛋白去乙醯化酶抑製劑(histone deacetylase inhibitor,HDACI),簡稱HDACIs,是一類化合物,有干擾與組蛋白去乙醯化酶的功能。組蛋白去乙醯化酶抑製劑通常可分為兩大類:NAD+-依賴性酶和Zn2+依賴性酶。Zn2+依賴性蛋白酶包括HDACsI、II、IV亞族;NAD+-依賴性酶主要是HDACsIII亞族。組蛋白去乙醯化酶抑製劑通過增加細胞內組蛋白的乙醯化程度,提高p21等基因的表達水平等途徑,抑制腫瘤細胞的增殖,誘導細胞分化和(或)凋亡。組蛋白去乙醯化酶抑製劑已成為腫瘤靶向治療的研究新熱點,其對腫瘤細胞遷移、侵襲、轉移的抑制作用和抗腫瘤血管生成作用也被證實。
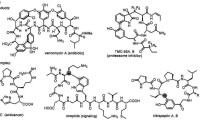
環肽類HDAC抑製劑
組蛋白去乙醯化酶抑製劑(HDACIs)包括結構不同的化合物,是一組有針對性的抗癌藥物。
當下發現的HDACi按其結構分為4類:
一、脂肪酸,如丁酸鹽、丁酸苯酯和丙戊酸,其中丙戊酸被用作抗癲癇藥物;
二、氧肟酸鹽,如TSA是被發現的第1個能抑制HDACs的天然氧肟酸,SAHA與TSA結構相似,是食品藥品管理局批准的第1個可以用於臨床的製劑;
三、環肽,如天然產物縮酚酸肽FK-228、apicidin和環氧肟酸;
四、苯醯胺類,如MS-275、MGCD0103。有些HDACi如TSA、SAHA屬非選擇性,可同時抑制Ⅰ類及Ⅱ類HDACs;有些HDACi屬於選擇性的,如MS-275對Ⅰ類HDACs的抑制作用強於Ⅲ類HDACs,而對HDAC6、HDAC8幾乎沒有影響。但是現階段對於決定不同HDACs活性的組蛋白特異序列還知之甚少。
IPF是一種慢性進展性、生存期短且病因不明的肺部疾病。肌成纖維細胞活化、增殖、分化是致纖維化的關鍵因素,轉化生長因子β1(TGF-β1)是主要的促纖維化因子。研究表明TGF-β1在體內外均可促進成纖維細胞分化為肌成纖維細胞(通過SMAD2、SMAD3磷酸化途徑)及上皮細胞轉化為間質成分(通過調節因子AKT磷酸化途徑),引起特徵性α-平滑肌肌動蛋白(α-SMA)、Ⅰ型膠原表達增加及纖維組織的收縮反應,而敲除HDAC4基因可阻斷TGF-β1介導的AKT磷酸化途徑。Guo等的實驗結果提示TSA主要通過下調HDAC4表達、抑制調節因子AKT磷酸化途徑,減少α-SMA及Ⅰ型膠原轉錄及表達,從而起到抗纖維化作用,但TSA無法抑制SMAD2及SMAD3磷酸化途徑。研究發現在環氧化物酶2(COX-2)作用下,前列腺素Ez(PGE2)可抑制肌成纖維細胞增殖及抑制膠原mRNA的轉錄,降低膠原水平而起到抗纖維化作用。Coward等的研究表明,與對照組相比,IPF患者肺成纖維細胞經TGF-β1(2ng/ml)處理后測得COX-2mRNA轉錄因子與啟動子結合能力受損,由HDAC1、HDAC2、HDAC3構成的相關轉錄遏阻,物複合體含量明顯增加,COX-2mRNA啟動子的組蛋白H3、H4去乙醯化酶含量明顯降低,COX-2mRNA水平明顯下降,PGE2含量也明顯減少。當IPF患者肺成纖維細胞同時經HDACi(10μmol/LSAHA或10nmol/LLBH589)及TGF-β1抑製劑處理后,COX-2mRNA啟動子的組蛋白H3、H4乙醯化酶明顯升高,可維持COX-2正常表達,減輕肺組織的纖維化程度。
腎間質纖維化以腎成纖維細胞異常活化及增生為特點,特徵性改變包括α-SMA表達及細胞外基質成分的增加。成纖維細胞的活化及增殖需要大量的生長因子及細胞因子如TGF、血小板衍生生長因子(PDGF)、成纖維細胞生長因子、白介素6(IL-6)等的參與。Kinugasa等的研究提示單側輸尿管梗阻致腎纖維化的小鼠模型中,給予小鼠FR276457(一種非特異性的HDACi,Ⅰ類HDAC及Ⅱ類HDACs抑製劑),連續14d每日鼻飼20mg/kg和40mg/kg,可抑制羥脯氨酸的生成及膠原α1mRNA的表達,同時體內和體外實驗均提示FR276457抑制促纖維化因子單核細胞趨化蛋白-1(MCP-1)的表達。Marumo等的研究提示在單側輸尿管梗阻致腎小管間質損傷的小鼠模型中,腹腔注射TSA10mg·kg·d,共2~5d,可抑制α-SMAmRNA及膠原α1mRNA的表達,同時通過抑制細胞因子如MCP-1、集落刺激因子-1進而減弱巨噬細胞滲入及早期腎小管的纖維化改變。Noh等的研究提示在大鼠的糖尿病腎臟模型中,TGF-β1促使HDAC2的活性明顯增高,進而造成細胞外基質的累積以及上皮一間質的轉化。而TSA(0.5mg·kg·d,共4周,腹腔內注射)通過抑制HDAC2的活性,在基因和蛋白的水平上均抑制了細胞外基質成分的表達,遏制了上皮間質的轉化。HDACs調節的腎成纖維細胞的活化通過信號轉導子和轉錄激活子3(STAT3)磷酸化途徑完成。研究顯示TSA可通過抑制STAT3磷酸化及核轉移阻斷相關信號途徑而減弱腎成纖維細胞增殖,起到抑制纖維化的作用。
心力衰竭分為以左心泵血功能受損為特徵的收縮功能衰竭和心肌舒張充盈功能異常為特徵的舒張功能衰竭。在病理基礎上表現為心肌過度肥大及細胞外基質沉積,Ⅰ型、Ⅲ型膠原累積,間質纖維化,心室擴張及心室壁變薄。因此有學者認為心肌過度肥大是心肌纖維化的一種早期表現。有研究顯示在壓力負荷所致心肌病變中轉錄因子心肌細胞增強因子-2的轉錄活性明顯增強,而Ⅱa類HDACs作用MEF-2靶基因形成複合體抑制其活性。除了通過上述直接作用阻礙基因表達外,Ⅱa類HDACs還通過與血清反應因子、活化T細胞核因子等間接關聯來抑制重構。因此,Ⅱa類HDACs被認為具有抑制心肌肥大、抑制心室重構進而抑制纖維化的作用。雖然研究表明Ⅱa類HDACs對心肌細胞具有抗纖維化作用,但是體內實驗證明非特異性HDACiTSA(Ⅰ類和Ⅱ類HDACs的抑製劑)可有效地抑制心肌細胞肥大。其中可能的解釋是Ⅰ類HDACs有促纖維化作用,且促纖維化作用程度大於Ⅱa類HDACs對心肌細胞的抗纖維化作用,因此非特異性HDACiTSA總體作用表現為抑制心肌肥大。雖然Ⅰ類HDACs的促纖維化作用機制尚不明確,但是多項研究提示Ⅰ類HDACs抗心肌肥大及纖維化與多種蛋白複合體的調控有關,其中包括組蛋白及一些轉錄因子的去乙醯化修飾。另外心房纖維化是房性心律失常的主要病因之一,Liu等的研究提示HDACs過度活化下調連接蛋白40,引起轉基因小鼠的心房纖維化,誘發房性心律失常。而給予小鼠腹腔內注入TSA0.6mg·kg·d持續14d,房性心律失常的持續時間縮短及心肌細胞自律性降低。因此HDACi的應用可為某些心臟疾病的治療帶來新的方法。
多個炎症性疾病動物模型的研究表明HDACi具有抗炎作用。HDACi除了通過調控組蛋白乙醯化狀態引起炎性細胞因子的表達,還可以通過調控包括轉錄因子在內的非組蛋白的乙醯化狀態,抑制或是激活炎性因子的表達,其中最主要的通過對核轉錄因子KB的抑制實現的。但是不同的HDACs具有不同的乙醯化模式,調節不同的基因。如系統性紅斑狼瘡MRL-Ipr/1pr小鼠模型,TSA和SAHA可下調炎性細胞因子IL-12和IL-6mRNA和蛋白的水平;在小鼠的哮喘模型中,TSA可以降低支氣管肺泡灌洗液中IL-4、IL-5和lgE的濃度,進而減緩哮喘發展。但也有關於HDACi能促進炎性細胞因子表達的報道。Suuronen等報道,在小膠質N9細胞TSA可以增強由脂多糖誘導的NO和IL-6的產生。因此,HDACi對於炎症反應的調控可以是基因轉錄激活劑,也可以是抑製劑。
丹麥科學家以期證明“找到可大範圍應用且不太昂貴的治癒HIV(人類免疫缺陷病毒)的方法是可能的”。
這種療法利用在治療癌症時更普遍使用的組蛋白去乙醯化酶活性抑製劑,來把HIV從病人的DNA中趕出來。丹麥的研究人員正在使用一種藥效尤其強大的組蛋白去乙醯化酶活性抑製劑帕比司他。
在用於惡性腫瘤的治療中,HDACi有效且耐受性好。正常細胞對高濃度的HDACi具有高耐受性,因此HDACi被認為是無毒性反應的,而且有實驗表明低劑量的HDACi在缺氧、炎性反應或負荷壓力作用下具有神經及腎臟保護作用。非特異性HDACi在腫瘤的Ⅰ期及Ⅱ期臨床試驗中被報道的不良反應有噁心、嘔吐、反應血液系統異常及QT間期延長,非特異性的HDACi如SAHA、LBH589、ITF-2357可能造成一過性的血小板減少或是骨髓抑制,而特異性的HDACi在用於腫瘤的研究中是否會引起相應的不良反應尚無定論,另外作為低劑量HDACi尚無不良反應的報道。因此,具有臟器保護作用且有更好的耐受性的低劑量的HDACi越來越多被用來嘗試治療慢性疾病。
HDACi在慢性纖維化性疾病中的作用引起越來越多的關注。HDACi通過對組蛋白及非組蛋白(轉錄因子等)修飾作用,多途徑抑制多種炎性因子和促纖維化因子而起到抑制炎症和纖維化作用。且HDACi所需的治療劑量低,耐受好,不良反應少,值得進一步深入研究。
一、廣州生物院組利用計算機輔助藥物設計原理和合理藥物設計的方法成功設計合成了新型組蛋白去乙醯化酶I型選擇性抑製劑。該先導化合物對HDAC1和HDAC9的選擇性是天然物largazole的10倍。這為進一步研究具有選擇性的組蛋白去乙醯化酶抑製劑提供了非常好的模型。該研究成果已於2012年12月在美國化學會期刊ACS Medicinal Chemistry Letters上在線發表(DOI:10.1021/ml300371t)。
組蛋白去乙醯化酶是抗腫瘤藥物的一個可靠靶點,具有選擇性的組蛋白去乙醯化酶抑製劑已經成為抗腫瘤藥物研究的熱門領域。該研究對腫瘤表觀遺傳及個體化治療提供了有益的方向。
該研究獲得國家自然科學基金和973項目的經費資助。
二、基因轉錄的有序調控是機體細胞維持正常功能的前提,如果基因轉錄調控功能紊亂,細胞就可能發生癌變。最近的研究發現,腫瘤的發生與核心組蛋白的乙醯化及去乙醯化的失衡有密切的關係。在有機體內,負責組蛋白乙醯化和去乙醯化的是一對功能相互拮抗的蛋白酶:組蛋白乙醯轉移酶(AHT)和組蛋白去乙醯酶(HDAC)。機體利用這兩種酶對組蛋白氮端氨基酸殘基進行乙醯化和去乙醯化,調節染色質的結構。進而調控基因轉錄。研究表明。組蛋白去乙醯酶抑製劑在體外和體內實驗中均能引起乙醯化核小體組蛋白的堆積,提高p21基因的表達水平,抑制腫瘤細胞的增殖。誘導細胞分化或凋亡,可用於多種惡性血液病及實體瘤的治療。科研人員對HDAC抑製劑的體內、外活性進行了系統的研究,部分HDAC抑製劑已經進入臨床研究階段。
三、核小體是真核生物染色質的基本單位,核小體的中部由4種組蛋白(H2A,H2B,H3,H4)各兩分子形成八聚體,周圍圍繞DNA,尾部為組蛋白H1。核小體表面修飾與基因表達調控聯繫密切。組蛋白乙醯化是其中一種重要的共價修飾,通過招募染色體構型重建複合體引起染色體構型發生改變。染色體構型改變使高度螺旋的染色質變得相對鬆弛,便於轉錄因子與DNA結合。使組蛋白乙醯化的主要是組蛋白乙醯轉移酶(histone acetyltransferase,HAT)和組蛋白去乙醯化酶抑製劑(histone deacetylase inhibitors,HDACI)。使組蛋白去乙醯化的酶稱為組蛋白去乙醯化酶(histone deacetylases,HDAC)。通常認為,組蛋白的高乙醯化是轉錄活躍的一個標誌,而低乙醯化則與轉錄抑制有關。
四、組蛋白是真核生物核染色體的重要組成成分。核心組蛋白的N端尾部可以通過各種修飾與DNA及其他蛋白髮生作用,調整核小體以及染色質結構,從而使被抑制的基因重新恢復活性。組蛋白乙醯化/去乙醯化修飾是基因轉錄調控的關鍵機制之一,這種修飾作用分別由組蛋白乙醯轉移酶(HAT)和組蛋白去乙醯化酶(HDACs)調控。HAT促使染色體的解聚,激活轉錄;而HDACs則封閉DNA,抑制轉錄過程。最近的多項研究表明,HAT/HDACs平衡的紊亂不僅會使基因表達失控,導致腫瘤的發生,而且與一些以慢性纖維化為主要特徵的疾病相關,如特發性肺纖維化(IPF)、慢性腎臟疾病、心力衰竭、系統性硬化等。因此,關於HDACs及組蛋白去乙醯化酶抑製劑(HDACi)在這些疾病病因及治療的研究受到越來越多的關注。
五、膀胱腫瘤是最常見的泌尿生殖系統腫瘤之一,其特點是易複發與轉移。降低膀胱腫瘤的複發與進展,提高膀胱腫瘤患者的生存時間與生活質量一直是臨床重要研究方向。組蛋白去乙醯化酶抑製劑作為新一代的抗腫瘤藥物,時下正成為腫瘤化學治療領域的熱點研究內容之一。研究組蛋白去乙醯化酶抑製劑在膀胱腫瘤中的作用機制將為指導臨床對膀胱癌的治療提供新的思路和方法。
六、神經膠質瘤(gliomas),是時下最常見的顱內惡性腫瘤,約佔全部顱內腫瘤的40%~50%。根據發生的部位和組織的不同,大致可分為以下幾類:星形細胞瘤(astrocytic)、室管膜瘤(ependymal)、少突膠質細胞瘤(oligodendroglial)、脈絡叢瘤(choroidplexus tumors)和多形性膠質母細胞瘤(gliobastoma)等。時下的治療手段主要集中於手術、放療和化療三個方面。由於該腫瘤多呈浸潤性生長,手術很難確切判定腫瘤邊界;放療對正常組織和細胞的損傷較大,且一些惡性膠質瘤(如惡性膠質母細胞瘤)在手術和放療后平均生存期不足12個月,2年生存率僅為10%。一些經典的膠質瘤化療藥物(如:卡莫司汀、尼莫司汀),由於人腦內血腦屏障(BBB)的存在及膠質瘤對這些藥物存在著耐藥性,治療效果也不十分明顯。因此,尋求有效的化療藥物,提高膠質瘤患者的生存率就成了時下一個重要研究目標。
基本信息
- 中文名
- 組蛋白去乙醯化酶抑製劑
- 外文名
- histone deacetylase inhibitor
- 別名
- HDACIs
- 藥品類別
- 抑製劑
- 適應症
- 治療腫瘤
- 是否處方葯
- 非處方葯