EST序列
EST序列
表達序列標籤(expressed sequence tags,ESTs)是一組短的、由大量隨機取出的 cDNA 庫克隆、經從 5' 或 3' 末端一次測序得到的組織或細胞基因組的一段 cDNA 序列, 一般長 60~500 bp。這一概念最早由 Venter於1991年提出。近年來由此形成的技術路線被廣泛應用於基因識別、繪製基因表達圖譜、尋找新基因等研究領域,並且取得了顯著成效。
At a time when the genomes of many species have been sequenced completely, a fundamental
resource expected by many researchers is a simple list of all of an organism's genes. A gene list,
together with associated physical reagents and electronic information, allows one to begin to
investigate the ways in which many genes interact in the complex system of the organism. However,
many species of medical and agricultural importance have not yet been prioritized for
genomic sequencing, and expressed cDNAs have provided the primary source of gene
sequences. Furthermore, when the genomic sequence of an organism becomes available, a collection
of cDNA sequences provides the best tool for identifying genes within the DNA sequence.
Thus, we can anticipate that the sequencing of transcribed products will remain a significant area
of interest well into the future.
The eara of high-throughput cDNA sequencing was initiated in 1991 by a landmark study from
Venter and his colleagues. The basic strategy involves selecting cDNA clones at random and
performing a single, automated, sequencing read from one or both ends of their inserts. They
introduced the term EST to refer to this new class of sequence, which is characterized by being
short (typically about 400–600 bases) and relatively inaccurate (around 2% error). The use of
single-pass sequencing was an important aspect of making the approach cost effective. In most
cases, there is no initial attempt to identify or characterize the clones. Instead, they are identified
using only the small bit of sequence data obtained, comparing it to the sequences of known genes and other ESTs. It is fully expected that many clones will be redundant with others already
sampled and that a smaller number will represent various sorts of contaminants or cloning artifacts.
There is little point in incurring the expense of high-quality sequencing until later in the
process, when clones can be validated and a non-redundant set selected.
Despite their fragmentary and inaccurate nature, ESTs were found to be an invaluable
resource for the discovery of new genes, particularly those involved in human disease processes. After the initial demonstration of the utility and cost effectiveness of the EST approach,
many similar projects were initiated, resulting in an ever-increasing number of human ESTs.
In addition, large-scale EST projects were launched for several other organisms of experimental
interest. In 1992, a database called dbEST was established to serve as a collection point for
ESTs, which are then distributed to the scientific community as the EST division of GenBank.
The EST division continues to dominate GenBank, accounting for roughly two-thirds of all submissions.
例
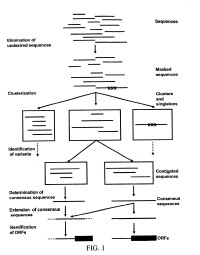
首先從樣品組織中提取mRNA ,在逆轉錄酶的作用下用oligo ( dT)作為引物進行RT -PCR合成cDNA ,再選擇合適的載體構建cDNA文庫,對各菌株加以整理,將每一個菌株的插入片段根據載體多克隆位點設計引物進行兩端一次性自動化測序,這就是EST序列的產生過程。
克隆全長cDNA序列的傳統途徑是採用噬斑原位雜交的方法篩選cDNA文庫,或採用PCR的方法,這些方法由於工作量大、耗時、耗材等缺點已滿足不了人類基因組時代迅猛發展的要求。而隨著人類基因組計劃的開展,在基因結構、定位、表達和功能研究等方面都積累了大量的數據,如何充分利用這些已有的數據資源,加速人類基因克隆研究,同時避免重複工作,節省開支,已成為一個急迫而富有挑戰性的課題擺在我們面前,採用生物信息學方法延伸表達序列標籤(ESTs)序列,獲得基因部分乃至全長cDNAycg,將為基因克隆和表達分析提供空前的動力,並為生物信息學功能的充分發揮提供廣闊的空間。
EST技術最常見的用途是基因識別,傳統的全基因組測序並不是發現基因最有效率的方法,這一方法顯得既昂貴又費時。因為基因組中只有2%的序列編碼蛋白質,因此一部分科學家支持首先對基因的轉錄產物進行大規模測序,即從真正編碼蛋白質的mRNA出發,構建各種cDNA文庫,並對庫中的克隆進行大規模測序。Adams等提出的表達序列標籤的概念標誌著大規模cDNA測序時代的到來。雖然ESTs序列數據對不精確,精確度最高為97%,但實踐證明EST技術可大大加速新基因的發現與研究。Medzhitov等通過果蠅黑胃TOLL蛋白進行dbEST資料庫檢索,該蛋白已證實在成熟果蠅抗真菌反應中發揮重要作用,通過同源分析的方法,找到相應的人類同源EST(登錄號為H48602),這為接下來研究人類TOLL同源蛋白的功能提供了很好的條件。hMSH5基因是從釀酒酵母菌MSH5存在30%的一致性,它與hMSH4特異性相互作用,在減數分裂和精子發生過程中發揮一定的作用。由此可見,應用EST技術,可以跳過生物分類學的界限,從生物模型的已識別基因迅速克隆出人和小鼠基因組相應的更複雜的未知基因。生物間在核苷酸水平上的進化差異阻礙了傳統意義上的雜交或以PCR為基礎的基因克隆策略,即使是親緣關係很接近的生物也不例外,如C.elegans和C.briggsae,它們僅在2~5千萬年前分化形成。而通過計算機進行dbEST進行資料庫篩選,其配製是電子雜交實驗,提供了一條更為廣泛的基因識別路線,這一路線允許基因組間存在差異,這使得基因識別與新基因克隆策略發生革命性變化,同時它也提供了一個足夠大小和複雜的基因資料庫,ESTs數量正以平均每月10萬條的速度遞增。
ESTs在多種以基因為基礎的人和植物基因組物理圖譜構建中扮演著重要角色。在這一應用中,從ESTs發展起來的PCR或雜交分析可用來識別YACs、BACs或其他含有大片段插入克隆類型的載體,它們是構建基因組物理圖譜的基礎,將EST與基因組物理圖譜相比較即可辨認出含有剩餘基因序列的基因組區間,包括調控基因表達的DNA控制元件,對這些元件進行分析就有可能獲得對基因功能的詳細了解。物理圖譜與遺傳圖譜間的相互參考,形成一個用途更廣泛的綜合資源,獲得這張綜合圖譜后,研究人員就可以孟德爾遺傳特徵為基礎,將相關基因定位在基因組區間上,並且通過查詢以ESTs為基礎的藶圖譜,即可獲得這一區間上所有基因的名單。該綜合資源用途的大小取決於EST資料庫中擁有的基因數目。人和小鼠EST的不斷擴充使其應用更加廣泛和便捷。
EST資料庫並非完美無瑕,因為ESTs不能被剪切為單列序列位點識讀,故精確度只能達到97%,另外,ESTS受制於表達傾向(expression bias),因為產生ESTs的cDNA是組織中豐富的mRNA以一定比例反轉錄而成,因此,表達水平很低的EST資料庫中很難找到,而表達量高的基因在EST資料庫中卻過量存在。雖然可在起始mRNA或由它合成雙鏈cDNA時進行富集,減小cDNA文庫,但cDNA文庫中仍存在大量高丰度的cDNA克隆。因此,一個理想的cDNA文庫必須去除或盡量消除多克隆的影響,這就涉及到cDNA文庫的前加工技術;均等化(normalization),減少與豐富編碼基因相關的cDNA數目;消減雜交(subtractive hybridization),應用序列標記cDNA識別並去除文庫中多餘的克降,這些技術的發展,使基因識別更依賴於EST技術,甚至可通過該技術獲得精確的基因組DNA序列,在華盛頓大學基因組測序中心和Sanger中心的聯合攻關下,C.elegans基因組10億個鹼基對的測序工作基本完成。因此ESTs是一系列基因尋找工具中不可缺少後部分,而這些工具都是基因組序列為基礎的。EST技術關於基因組DNA序列的其他應用還包括對基因內含子、外顯子排列的精確預測,選擇性接合事件的識別,反常基因組排列結構的識別等。
4、
利用計算機來協助克隆基因,稱為“電子”基因克隆(sillcon cloning),是與定位克隆、定位候選克隆策略並列的方法之一,即採用生物信息學的方法延伸EST序列,以獲得基因部分乃至全長的cDNA序列。EST資料庫的迅速擴張,已經並將繼續導致識別與克隆新基因策略發生革命性變化。
4.1
EST序列的獲取
利用計算機來協助克隆的第一步是必須獲得感興趣的EST,在dbEST資料庫中找出EST的最有途徑是尋找同源序列,標準:長度≥100bp,同源性50%以上、85%以下。可通過數個萬維網界而使用BLAST檢索程度實現,其中最常用的如NCBI(National Center for Biotechnology Information)的GenBank、義大利Tigem的ESTmachine(包括EST提取者和EST組裝機器)、THC(Tentative Human Consensus Sequences)資料庫、ESTBlast檢索程序——通過英國人類基因組作圖項目資源中心(Human Genome Mapping Project Resource Center,HGMP—RC)伺服器上訪問。然後將檢出序列組裝為重疊群(contig),以此重疊群為被檢序列,重複進行BLAST檢索與序列組裝,延伸重疊樣系列,重複以上過程,直到沒有更多的重疊EST檢出或者說重疊群序列不能繼續延伸,有時可獲得全長的基因編碼序列。獲得這些EST序列數據后,再與GeneBank核酸資料庫進行相似性檢測,假如鳳有精確匹配基因,將EST序列數據據EST六種閱讀框翻譯成蛋白質,接著與蛋白質序列資料庫進行比較分析。基因分析的結果大致有三種:第一是已知基因,是研究對象為人類已鑒定和了解的基因;第二是以前未經鑒定的新基因;第三是未知基因,這部分基因之間無同種或異種基因的匹配。新基因和未知基因將進一步用於生物學研究。
4.2
基因的電子定位
基因的電子定位採用NCBI的電子PCR程序進行檢索,尋找EST序列上是否存在序列標籤位點(sequence tagged sites,STS),STS作為基因組中的單拷貝序列,是新一代的遺傳標記系統,其數目多,覆蓋密度較大,達到平均每1kb一個STS或更密集。將尋找到的STS與相應的染色體相比較,即可將此序列定位在該染色體上。
4.3
IMAGE克隆的索取
許多ESTs所對應的cDNA克隆可通過基因組及其表達的整合分子分析(intergrated molecular analysis of genomes and their expression,IMAGE)協定免疫索取,這與電子基因克隆相輔相成,IMAGE協定由美國LLNL國家實驗室主持,宗旨是共享排列好的cDNA文庫中的克隆重,大規模的EST測序項目如Merk&Cow公司投資的人類ESTs項目等都加入了IMAGE協定。當研究者通過另外的途徑得到基因的部分序列,並通過同源性檢索后發現該片段與加入IMAGE協定的EST序列高度同源時,便可免費索取其原始克隆,可通過美國的ATCC組織(American Type Culture Collection)索取,從而避免或減輕篩選全長基因的麻煩,以集中精力進行基因的功能研究。
EST作為表達基因所在區域的分子標籤因編碼DNA序列高度保守而具有自身的特殊性質,與來自非表達序列的標記(如AFLP、RAPD、SSR等)相比更可能穿越家系與種的限制,因此EST標記在親緣關係較遠的物種間比較基因組連鎖圖和比較質量性狀信息是特別有用。同樣,對於一個DNA序列缺乏的目標物種,來源於其他物種的EST也能用於該物種有益基因的遺傳作圖,加速物種間相關信息的迅速轉化。具體說,EST的作用表現在: (1)用於構建基因組的遺傳圖譜與物理圖譜; (2)作為探針用於放射性雜交; (3)用於定位克隆; (4)藉以尋找新的基因; (5)作為分子標記; (6)用於研究生物群體多態性; (7)用於研究基因的功能; (8)有助於藥物的開發、品種的改良; (9)促進基因晶元的發展等方面。正是因為EST表現出了這些巨大潛能,使其得到了充分的利用與發展。