腎上腺髓質腺瘤
可引起心腦血管意外而危及病人生命
腎上腺髓質腺瘤是發生於腎上腺髓質、交感神經節、旁交感神經節或其他部位的嗜鉻組織中的腫瘤。這種腫瘤持續或間接地釋放大量兒茶酚胺(去甲腎上腺素、腎上腺素、多巴胺)引起發作性高血壓伴交感神經興奮為主要臨床表現的內分泌疾病。嚴重發作可引起心腦血管意外而危及病人生命。若能早期診斷,良性腎上腺髓質腺瘤經手術可以治癒。
本病是一種罕見的繼發性高血壓,患病率占高血壓病的0.1%~1%。近年來隨著醫學的發展和醫生警惕性的提高,本病的發現已漸漸增多,男女發病數相似,各年齡組均可發生,以20 ~50歲最多見,兒童病人也不少。小兒患者男女比例為2∶1。本病可有家族史,稱為家族性腎上腺髓質腺瘤,屬於多發性內分泌腺瘤中的Ⅱ、Ⅲ型,良性者約佔80%~90%,惡性者佔10%~2 0%。腫瘤直徑約12~16厘米,重量自數克至3公斤,大小不等,一般均在100克左右。
引起腎上腺髓質功能亢進的主要原因有嗜鉻細胞瘤、惡性嗜鉻細胞瘤和腎上腺髓質增生。
嗜鉻細胞呈圓形或橢圓形,有完整包膜,周圍血管豐富怒張。腫瘤一般較大,直徑在2~6cm。約90%發生在腎上腺髓質,其餘10%可發生在腎上腺以外的部位。發生在腎上腺以外的腫瘤,多見於腹膜後主動脈旁,包括嗜鉻體(Zuckerkandl體)。也可在腎臟、腎門、肝門、胰頭附近,脾、腹腔動脈附近,髂血管旁,卵巢、膀胱區。腹腔以外的嗜鉻細胞瘤極少見,如后縱隔脊柱旁,偶見於頸部、顱內及睾丸內。腎上腺嗜鉻細胞瘤90%是單發,雙側或多發佔10%。腫瘤一般屬良性(佔90%左右),切面呈桔黃色,常見出血、壞死及囊樣變,血管豐富。間質很少,腫瘤細胞較大,為不規則多角形。胞漿中顆粒較多,與正常腎上腺髓質細胞相似,但較大。鉻酸鹽可使顆粒著色,故稱嗜鉻細胞瘤。約10%的嗜鉻細胞瘤為惡性,但單從組織形態學上有時難以鑒別,其主要表現在其有惡性行為,即腫瘤包膜浸潤,淋巴、肝、骨和肺等臟器的轉移。
極少數病例的臨床表現和生化檢查均符合嗜鉻細胞瘤的診斷,但無腫瘤存在,是腎上腺髓質增生所致。增生的髓質細胞在形態上與正常髓質細胞無區別,僅見整個腎上腺體積較大,腺體飽滿,表面隆起,髓質層增寬(與皮質比大於1:10)。髓質增生為雙側性病變,但兩側的增生程度可有差異。
臨床表現是由於腫瘤細胞分泌大量兒茶酚胺所致。腎上腺嗜鉻細胞瘤主要分泌腎上腺素。而腎上腺外部位的嗜鉻細胞瘤主要分泌去甲腎上腺素,這是由於異位的腫瘤組織中缺乏甲基轉移酶之故。嗜鉻細胞瘤分泌兒茶酚胺可以是間歇性的也可以是持續性的,從而出現了多變的臨床癥狀。
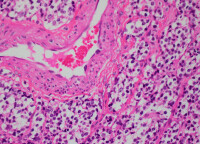
神經母細胞瘤
1964年Pear將廣泛存在於內分泌腺體及其它組織中能產生內分泌多肽等物質的細胞統稱為APUD細胞(amine precursor uptake and decarboxylation cells),這些細胞均起源於神經嵴。例如垂體分泌ACTH、促黑色素的細胞,胰島分泌胰高血糖素、胰島素和胃泌素的α,β及δ細胞,消化道分泌胃泌素的G細胞,甲狀腺分泌降鈣素的C細胞,腎上腺髓質的嗜鉻細胞,以及肺支氣管分泌肺血管活性肽的細胞等。當神經嵴細胞發育異常時,就可以出現多發性內分泌腺瘤(mutiple endocrine neoplasia,MEN)。其引起的病症稱多發性內分泌腺瘤綜合征(MENS)。Wermer認為這是一組以常染色體顯性遺傳為特徵的遺傳性疾病,具有較高的外顯率。根據臨床表現的不同,可以分成①MENⅠ型(Wermer綜合症):包括甲旁亢,胰島性低血糖,消化道潰瘍(zollinger-Ellison綜合症)。可伴有垂體前葉或腎上腺皮質功能亢進。②MENⅡ型(Sipple綜合症):又可分成Ⅱa型(甲狀腺髓樣癌、嗜鉻細胞瘤)和Ⅱb型(甲狀腺髓樣癌、甲旁亢、嗜鉻細胞瘤、其它多發性神經瘤、粘膜神經瘤、先天性巨結腸)。有人把伴有多發性粘膜神經瘤的Ⅱ型稱之為MENⅢ型。故凡嗜鉻細胞瘤伴有其它內分泌紊亂者應疑及MENSⅡ型可能。MENⅡ型中的甲狀腺髓樣癌起源於甲狀腺腺泡間的C細胞,能大量分泌降鈣素,使血鈣降低,從而刺激甲狀旁腺引起甲狀旁腺增生或腺瘤。故一般認為其甲旁亢是繼發性的。偶爾可產生前列腺素和ACTH,導致腹瀉、脂肪痢及皮質醇症。MENSⅡ型病例的腎上腺嗜鉻細胞瘤大多為雙側性並有惡性傾向。故此類病例應普查有血統關係的家庭其他成員,病人應長期密切隨訪。
部分嗜鉻細胞瘤有遺傳的特點
一部分嗜鉻細胞瘤有家庭遺傳傾向,其特點是:
①其發病年齡比非家庭性的為早。
②雙側發病率高達47%。
③在一個家庭成員中發病的成員,其發病時的年齡和腫瘤部位往往相同。
④常伴有多發性內分泌腫瘤Ⅱ型或神經外胚層發育異常。如多發性神經纖維瘤病、結節性硬化症、腦三叉神經多發性血管瘤等,應予注意。
最突出表現是高血壓,可以是陣發性(45%),或持續性(50%)和罕見缺乏高血壓(5%).約1/1000高血壓病人是腎上腺髓質腺瘤。高血壓是由於分泌一種或一種以上兒茶酚胺激素或前體:正腎上腺素,腎上腺素,多巴胺或多巴。常見的癥狀和體征是心動過速,多汗,體位性低血壓,呼吸加速,潮紅,冷和濕冷皮膚,嚴重頭痛,心絞痛,心悸,噁心,嘔吐,上腹部疼痛,視力障礙,氣急,感覺異常,便秘和瀕臨死亡感。可因捫及腫瘤,體位改變,腹部壓迫或按摩,誘導麻醉,情緒傷害,β-阻滯劑等促發陣發性發作,如腫瘤在膀胱可因排尿而誘發。
體檢,除常見高血壓外,一般正常,除非在發作期間。視網膜病和心臟擴大的嚴重性常常少於高血壓可預見的程度
鑒別診斷與病理學
需與腎上腺皮質增生鑒別。
嗜鉻細胞腫瘤分泌兒茶酚胺致使高血壓。
約80%腎上腺髓質腺瘤見於腎上腺髓質,但同樣可見於來自神經脊細胞的其他組織(見下文病理學).腎上腺髓質腫瘤兩性相等,雙側10%(兒童20%),通常良性(95%).腎上腺外腫瘤惡性多見(30%).雖然腎上腺髓質腺瘤可見於任何年齡,但最大發生率介於20~50歲。
腎上腺髓質腺瘤大小不同,但平均直徑僅5~6cm,通常重量50~200g,但數千克已見報道。罕見有大到足以捫及或引起壓迫或梗阻的癥狀。腫瘤通常包膜完整,顯微鏡下細胞形態似惡性。這些細胞有許多怪異外形伴有緻密,大或多核。不管組織學形態如何,假如腫瘤未侵犯包膜或轉移,可以看作為良性。除了腎上腺,腫瘤可見於腹膜后沿著主動脈旁交感神經鏈的嗜鉻組織,頸動脈體,楚克坎德爾體(Zuckerkandl,位於主動脈分叉),腸胃-泌尿系統,腦,心包和皮樣囊腫里。
腎上腺髓質腺瘤是家族性多內分泌腫瘤---ⅡA型(賽普爾綜合征)組成部分,可伴有甲狀腺髓樣癌和甲狀旁腺腺瘤(參見第10節).Ⅲ型綜合征已有描述,包括腎上腺髓質腺瘤,粘膜(口腔和眼)神經瘤和甲狀腺髓樣癌。與多發性神經纖維瘤(vonRecklinghausen病)明顯伴隨(10%),可以發現血管瘤,如在vonHippel-Lindau病中一樣。
大的腎上腺髓質腺瘤在上可見腎上極有軟組織腫塊影.小的腫瘤則不能發現。
可見腎上腺區有低或等回聲的腫塊、內部回聲細小均勻,圓或橢圓形,有包膜易被發現。若瘤內部有出血、囊變、壞死,則回聲不均勻。
平掃為密度均勻的腫塊,密度略低於肝,大的腫塊內可出現出血、囊變或壞死的低密度區。增強掃描腫瘤迅速強化,密度高於皮質腺瘤,甚為明顯。有時可發現雙側腎上腺髓質腺瘤。惡性腎上腺髓質腺瘤表現如其他腎上腺的惡性腫瘤,有時可發現肝臟或(和)淋巴結轉移徵象。臨床若有典型的腎上腺髓質腺瘤癥狀,化驗檢查亦符合,而腎上腺未見病變時,應再查胸、腹、盆腔,以發現異位的腎上腺髓質腺瘤。此外腎上腺髓質增生也會引起腎上腺髓質腺瘤的癥狀,此時與腎上腺皮質增生在影像學表現相似。
基本信息
- 中文名
- 腎上腺髓質腺瘤
- 別名
- 腎上腺腺瘤
- 癥狀表現
- 發作性高血壓伴交感神經興奮為主要臨床表現交感神經
- 併發症
- 可引起心腦血管意外而危及病人生命
- 就診科室
- 外科