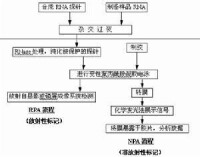
分子雜交
分子雜交
不同的DNA 片段之間,DNA 片段與RNA 片段之間,如果彼此間的核苷酸排列順序互補也可以復性,形成新的雙螺旋結構。這種按照互補鹼基配對而使不完全互補的兩條多核苷酸相互結合的過程稱為分子雜交。
分子雜交(molecular hybridization)確定單鏈核酸鹼基序列的技術。其基本原理是待測單鏈核酸與已知序列的單鏈核酸(叫做探針)間通過鹼基配對形成可檢出的雙螺旋片段。這種技術可在DNA與DNA,RNA與RNA,或DNA與RNA之間進行,形成DNA-DNA,RNA-RNA或RNA-DNA等不同類型的雜交分子。
作為探針的已知DNA或RNA片段一般為30~50核苷酸長,可用化學方法合成或者直接利用從特定細胞中提取的mRNA。探針必須預先標記以便檢出雜交分子。標記方法有多種,常用的為同位素標記法和生物素標記法。雜交方法又可分為液相雜交和固相雜交。
使單鏈DNA或 RNA分子與具有互補鹼基的另一DNA或RNA 片斷結合成雙鏈的技術。即核酸分子間的雜交。相互雜交的兩種核酸分子間有時並非完全一致,但必有一定的相關性。早在1961年有人創建了DNA與RNA間的分子雜交,但至70年代才發展成基因分析中一種重要的分析技術,可鑒定基因的特異性。例如可將具有一定已知順序的某基因的 DNA片段,標上放射性核素,構成核酸探針,將它通過分子雜交與缺陷的基因結合,產生雜交信號,從而把缺陷的基因顯示出來,藉此可對許多遺傳性疾病進行產前診斷。現又用核酸分子雜交技術診斷乙型肝炎,以及研究其他病毒性疾病和癌瘤的基因結構(癌基因)等。
核酸分子雜交的方法並不複雜。在雜交前先把待分析的DNA加熱或加鹼使之變性,分開成單鏈;然後加入放射性核素標記的核酸探針,降溫退火,即獲帶有放射性核素標記的雙鏈雜交分子。1979年又發展成轉膜雜交,即將電泳分開的核酸片段,經過轉移電泳轉移到硝酸纖維素膜上,進行固相雜交反應,用放射自顯影檢出之。此即著名的薩瑟恩氏印跡法。
無庸置疑,分子雜交的基礎是兩個核酸分子間的完全一致,或有一定的相似性或相關性。若所用的核酸探針的片段較長(幾百個核苷酸以上),可以得到很準確的鑒定結果。但若人工合成的寡核苷酸探針片段較短(如20~30個核苷酸),則難免會出現準確性差的假陽性現象。現又有非放射性核素標記核酸探針,如以鹼性磷酸酶標記的酶標核酸探針,以及以生物素標記的核酸探針等,這必將有助於推動分子雜交在臨床醫學中的廣泛應用。
分子雜交
分子雜交(molecular hybridization)不同來源的核酸單鏈之間或蛋白質亞基之間由於結構互補而發生的非共價鍵的結合。根據這一原理髮展起來的各種技術統稱為分子雜交技術,核酸分子雜交技術是分子遺傳學中的重要研究方法。
核酸分子雜交具有互補的鹼基順序的單鏈核酸分子,可以通過鹼基對之間非共價鍵(主要是氫鍵)的形成即能出現穩定的雙鏈區,這是核酸分子雜交的基礎。雜種分子的形成並不要求兩條單鏈的鹼基順序完全互補,所以不同來源的核酸單鏈只要彼此之間有一定程度的互補順序(即某種程度的同源性)就可以形成雜種雙鏈。使單鏈聚合為雙鏈的過程稱為退火或復性。用分子雜交進行定性或定量分析的最有效方法是將一種核酸單鏈用同位素標記成為探針,再與另一種核酸單鏈進行分子雜交。由於同位素被檢出的靈敏度高,所以在兩種核酸分子之間即使只有百萬分之一的同源順序也可以彼檢出。
核酸分子雜交按雜交中單鏈核酸所處的狀態可分為液相、固相和原位3類。前二類方法都要求預先從細胞中分離並純化雜交用的核酸。在液相分子雜交中,兩種來源的核酸分子都處於溶液中,可以自由運動,其中有一種常是用同位素標記的。從復性動力學數據的分析可探知真核生物基因組結構的大致情況,如各類重複順序的含量及分佈情況等。在固相分子雜交中,一種核酸分子被固定在不溶性的介質上,另一種核酸分子則處在溶液中,兩種介質中的核酸分子可以自由接觸。常用的介質有硝酸纖維素濾膜、羥基磷灰石柱、瓊脂和聚丙烯醯胺凝膠等。早期用瓊脂作為固定介質的分子雜交方法曾被用來測定從細菌到人多種生物的DNA的同源程度。
1975年由E.薩瑟恩首創的薩瑟恩吸印法是一種方便易行的固相雜交方法。先將待測的DNA用限制性內切酶切成片段,然後通過凝膠電泳把大小不同的片段分開,再把這些DNA片段吸印到硝酸纖維膜上,並使吸附在濾膜上的DNA分子變性,然後和預先製備的DNA或RNA探針進行分子雜交,最後通過放射自顯影就可以鑒別待測的哪個DNA片段和探針具有同源順序。精確控制雜交及洗滌條件(如溫度及鹽濃度),在同源順序中即使有一對鹼基錯配也可檢出。這技術已應用於遺傳病(基因病)的臨床診斷。另一種相似的方法是將待測的RNA片段吸印在重氮苄氧甲基(DBM)纖維素膜或濾紙上,然後和預先製備的DNA探針進行分子雜交,這種方法稱為諾森吸印法。這些技術大大地推進了分子遺傳學研究和重組DNA技術的應用。
原位(分子)雜交實際上是固相雜交的另一種形式,雜交中的一種DNA處在未經抽提的染色體上,並在原來位置上被變性成單鏈,再和探針進行分子雜交。在原位雜交中所使用的探針必須用比活性高的同位素標記。雜交的結果可用放射自顯影來顯示,出現銀粒的地方就是與探針互補的順序所在的位置。

分子雜交理論和技術
在基因定位工作中,還可以應用一種在電鏡下直接觀察雜種分子的原位雜交方法,即所謂的R-環法。R-環是在雙鏈DNA分子部分變性的情況下,單鏈的RNA分子和相應的DNA序列中的一條互補的單鏈形成RNA-DNA的雜合雙鏈,同時DNA的另一條單鏈向外突出而形成的環狀圖象。由於每種mRNA由特定的基因所轉錄,和這一基因的一個DNA單鏈具有互補結構,因此通過觀察標記mRNA參與構成的R環的位置就可以對產生這種mRNA的基因進行定位,這一方法還可以用來研究斷裂基因的結構。
在重組DNA技術中,有些專門的原位雜交方法可以用來篩選有探針順序的重組質粒或噬菌體。此外,分子雜交方法還可以用來分離具有特定順序的核酸分子。例如使探針與細胞的總DNA進行分子雜交,利用羥基磷灰石柱只吸附雙鏈分子的特性,可以分離與探針互補的DNA片段或RNA分子。
蛋白質分子雜交來自不同的蛋白質的亞基間也可以形成非共價鍵的結合,這是蛋白質分子雜交的基礎。蛋白質分子雜交技術可以用來研究蛋白質的結構和功能以及相似的蛋白質(如同工酶、血紅蛋白等)的親緣關係。例如通過人、狗、兔、驢、小鼠和蛙等動物的血紅蛋白的蛋白質分子雜交測驗,根據能否形成雜種分子和雜種分子的量,可以判斷它們的蛋白質分子的相似程度。
分子雜交是通過配對鹼基對之間的非共價鍵(主要是氫鍵)結合,從而形成穩定的雙鏈區。雜交分子的形成並不要求兩條單鏈的鹼基順序完全互補,所以不同來源的核酸單鏈只要彼此之間有一定程度的互補順序(即某種程度的同源性)就可以形成雜交雙鏈。分子雜交可在DNA與DNA、RNA與RNA或RNA與DNA的二條單鏈之間進行。由於DNA一般都以雙鏈形式存在,因此在進行分子雜交時,應先將雙鏈DNA分子解聚成為單鏈,這一過程稱為變性,一般通過加熱或提高pH值來實現。用分子雜交進行定性或定量分析的最有效方法是將一種核酸單鏈用同位素或非同位素標記成為探針,再與另一種核酸單鏈進行分子雜交。
雜交類型
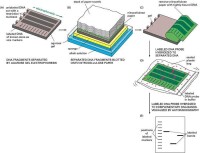
DNA分子雜交
將參加反應的一條核酸鏈先固定在固體支持物上,一條反應核酸遊離在溶液中。固體支持物有硝酸纖維素濾膜、尼龍膜、乳膠顆粒、磁珠和微孔板等。由於固相雜交后,未雜交的遊離片段可容易地漂洗除去,膜上留下的雜交物容易檢測和能防止靶DNA自我復性等優點,所以最為常用。常用的固相雜交類型有:菌落原位雜交、斑點雜交、狹縫雜交、Southern印跡雜交、Northern印跡雜交、組織原位雜交和夾心雜交等。
液相雜交
所參加反應的兩條核酸鏈都遊離在溶液中,一種研究最早且操作複合的雜交類型,在過去的30年裡雖有時被應用,但總不如固相雜交那樣普遍。主要缺點是雜交後過量的未雜交探針在溶液中除去較為困難和誤差較高。近幾年由於雜交檢測技術的不斷改進,商業性基因探針診斷盒的實際應用,推動了液相雜交技術的迅速發展。
固相膜核酸分子雜交
菌落原位雜交
(Colony in situ hybridization)
將細菌從培養平板轉移到硝酸纖維素濾膜上,然後將濾膜上的菌落裂菌以釋出DNA,再烘乾固定DNA於膜上,與32P標記的探針雜交,放射自顯影檢測菌落雜交信號,並與平板上的菌落對位。
斑點雜交
(Dot blotting)
該方法是將被檢標本點到膜上,烘烤固定。這種方法耗時短,可做半定量分析,一張膜上可同時檢測多個樣品。為使點樣準確方便,市售有多種多管吸印儀(manifolds),如MinifoldI和II、Bio-Dot(Bio-Rad)和Hybri-Dot,它們有許多孔,樣品加到孔中,在負壓下就會流到膜上呈斑點狀或狹縫狀,反覆沖洗進樣孔,取出膜烤乾或紫外線照射以固定標本,這時的膜就可以進行雜交。
Southern印跡雜交
(Southern blotting)
是研究DNA圖譜的基本技術,在遺傳診斷DNA圖譜分析及PCR產物分析等方面有重要價值。基本方法是將DNA標本用限制性內切酶消化后,經瓊脂糖凝膠電泳分離各酶解片段,然後經鹼變性,Tris緩衝液中和,高鹽下通過毛吸作用將DNA從凝膠中轉印至硝酸纖維素濾膜(尼龍膜也較長用)上,烘乾固定后即可用於雜交。凝膠中DNA片段的相對位置轉移到濾膜的過程中繼續保持著。附著在濾膜上的DNA與32P標記的探針雜交,利用放射自顯影術確定探針互補的每條DNA帶的位置,從而可以確定在眾多酶解產物中含某一特定序列的DNA片段的位置和大小。
Northern印跡雜交
(Northern blotting)
DNA印跡技術由Southern於1975年創建,稱為Southern印跡技術。RNA印跡技術正好與DNA相對應,故被稱為Northern印跡雜交,與此原理相似的蛋白質印跡技術則被稱為westernblotting。Northern印跡雜交的RNA吸印與Southern印跡雜交的DNA吸印方法類似,只是在進樣前用甲基氧化汞、乙二醛或甲醛使RNA變性,而不用NaOH,因為它會水解RNA的2’-羥基基團。RNA變性後有利於在轉印過程中與硝酸纖維素膜結合,它同樣可在高鹽中進行轉印,但在烘烤前與膜結合得並不牢固,所以在轉印后不能用低鹽緩衝液洗膜,否則RNA會被洗脫。在膠中不能加EB,因它為影響RNA與硝酸纖維素膜的結合,為測定片段大小,可在同一塊膠上加標記物一同電泳,之後將標記物膠切下,上色、照像。樣品膠則進行Northern轉印,標記物膠上色的方法是在暗室中將其浸在含5μg/mlEB的0.1mol/L醋酸銨中10min,在水中就可脫色。在紫外光下用一次成像相機拍照時,上色的RNA膠要儘可能少接觸紫外光,若接觸太多或白熾燈下暴露過久,會使RNA信號降低。從瓊脂糖凝膠中分離功能完整的mRNA時,甲基氫氧化汞是一種強力、可逆變性劑,但是有毒,因而許多人喜用甲醛作為變性劑。所有操作均應避免RNase的污染。
組織原位雜交
(Tissue in situ hybridization)
簡稱原位雜交,指組織或細胞的原位雜交,它與菌落的原位雜交不同,菌落原位雜交需裂解細菌釋出DNA,然後進行雜交。而原位雜交是經適當處理后,使細胞通透性增加,讓探針進入細胞內與DNA或RNA雜交。因此原位雜交可以確定探針的互補序列在胞內的空間位置,這一點具有重要的生物學和病理學意義。例如,對緻密染色體DNA的原位雜交可用於顯示特定的序列的位置;對分裂期間核DNA的雜交可研究特定序列在染色質內的功能排布;與細胞RNA的雜交可精確分析任何一種RNA在細胞中和組織中的分佈。此外,原位雜交還是顯示細胞亞群分佈和動向及病原微生物存在方式和部位的一種重要技術。
用於原位雜交的探針可以是單鏈或雙鏈DNA,也可以是RNA探針,探針的長度通常以100~400nt為宜,過長則雜交效率減低。2013年研究結果表明,寡核苷酸探針(16~30nt)能自由出入細菌和組織細胞壁,雜交效率明顯高於長探針。因此,寡核苷酸探針和不對稱PCR標記的小DNA探針或體外轉錄標記的RNA探針是組織原位雜交的優選探針。
試驗方法
總述:
通過鹼基對之間非共價鍵的形成即出現穩定的雙鏈區,這是核酸分子雜交的基礎。使單鏈聚合雙鏈的過程稱為退火或復性。核酸雜交技術基本上是Hall等1961年的工作開始的,探針與靶序列在溶液中雜交,通過平衡密度梯度離心分離雜交體。該法很慢、費力且不精確,但它開拓了核酸雜交技術的研究。Bolton等1962年設計了第一種簡單的固相雜交方法,稱為DNA-瓊脂技術。變性DNA固定在瓊脂中,DNA不能復性,但能與其它互補核酸序列雜交。典型的反應是用放射性標記的短DAN或RNA分子與膠中DNA雜交過夜,然後將膠置於柱中進行漂洗,去除遊離探針,在高溫、低鹽條件下將結合的探針洗脫,洗脫液的放射性與結合的探針量呈正比。該法尤其適用於過量探針的飽和雜交實驗。60年代末,Britten等設計了另一種分析細胞基因組的方法。該法是研究液相中DNA的復性以比較不同來源核酸的複雜度,典型的方法是:從不同生物體(細菌、酵母、魚和哺乳動物等)內分離DNA,用水壓器剪切成長約450核苷酸(nt)的片段。剪切的DNA液(含0.12mol/L磷酸鹽緩衝液或0.18mol/LNa+),經煮沸使dsDNA熱變性成ssDNA。然後冷至約60℃,在此溫度孵育過程中,測定溶液一定時間內的UV260nm的吸光度(減色效應)來監測互補鏈的復性程度。通常該實驗可比較不同來源生物DNA的復性速率,並可建立序列複雜度與動力學複雜度間的關係。
一60年代中期Nygaard等的研究為應用標記DNA或RNA探針檢測固定在硝酸纖維素(NC)膜上的DNA序列奠定了基礎。如Brown等應用這一技術評估了爪蟾rRNA基因的拷貝數。RNA在代謝過程中被3H尿嘧啶標記,並在過量的情況下與膜上固定的基因組DNA雜交,繼而用RNase處理,消化非特異性結合的RNA。漂洗後計數以測定雜交探針的量。通過計算與已知量DNA雜交的RNA量即可評估rRNA基因數。由於當時缺乏特異探針,這種方法不能用於研究其它特異基因的表達,這些早期過量探針膜雜交試驗實際上是現代膜雜交實驗的基礎。進入70年代早期,許多重要的發展促進了核酸雜交技術的進展。例如,對特異基因轉錄產物的分析和對動力學雜交實驗又有興趣。固相化的PolyU–Sepharose和寡(dT)-纖維素使人們能從總RNA中分離PolyA+RNA。用mRNA的經純化技術可從網織紅細胞總RNA中製備α-和β-珠蛋白mRNA混合物。這些珠蛋白mRNA首次被用於合成特異的探針以分析珠蛋白基因的表達。由於製備cDNA探針很繁瑣,所獲得cDNA的長度和純度也不穩定。所以尋求新的探針來源是使分子雜交技術進一步推廣的基礎。
70年代末期到80年代早期,分子生物學技術有了突破性進展,限制性內切酶的發展和應用使分子克隆成為可能。各種載體系統的誕生,尤其是質粒和噬菌體DAN載體的構建,使特異性DNA探針的來源變得十分豐富。人們可以從基因組DNA文庫和cDNA文庫中獲得特定基因克隆,只需培養細菌,便可提取大量的探針DNA。迄今為止,已克隆和定性了許多特異DNA探針。
由於固相化學技術和核酸自動合成儀的誕生,印跡技術,用數微克DNA就可分析特異基因。特異DNA或RNA序列的量和大小均可用Southern印跡和Northern印跡來測定,與以前的技術相比,大大提高了雜交水平和可信度。
儘管取得了上述重大進展,但分子雜交技術在臨床實用中仍存在不少問題,必須提高檢測單拷貝基因的敏感性,用非放射性物質代替放射性同位素標記探針以及簡化實驗操作和縮短雜交時間,這樣,就需要在以下三方面著手研究:第一,完善非放射性標記探針;第二,靶序列和探針的擴增以及信號的放大;第三,發展簡單的雜交方式,只有這樣,才能使DNA探針實驗做到簡便、快速、低廉和安全。
探針-靶反應
分子雜交實驗
核苷酸經某一原子、功能基團或長側鏈修飾后仍可能進行鹼基配對,這取決於修飾的部位和修飾的性質。這一特性有助於理解非放射性核酸探針標記物的設計和125I與DNA探針的化學結合。能與核酸結合的單一原子有銀、溴和碘等,這些元素可與嘧啶(胸腺嘧啶除外)環的C-5位或嘌呤環的C-8位反應而不影響氫鍵的形成。溴亦可與胸腺嘧啶的C-6位結合。而胞嘧啶的C-4和腺嘌呤的N-6就不能被修飾,否則會影響鹼基配對,儘管C的N-4位和A的N-6位參與了氫鍵形成,但它們也是標記位點。這是因為標記的探針每1kb只摻入10~30個修飾鹼基,即僅4%~12%的單個鹼基被修飾的類似物取代了。儘管摻入位點處的鹼基配對較弱或不存在,但對整個雜交分子的穩定性影響很小。防止氫鍵破壞的一種方法就是修飾探針,即探針克隆入M13載體中,只修飾載體區而不修飾插入片段。當用放射性同位素32P和35S標記核酸時,由於同位素是摻入核酸骨架的磷酸二脂鍵中,因此鹼基未發生任何修飾。在5’端的磷酸基團上可進行化學修飾,這是標記寡核苷酸探針的有效方法。因為這種方法是在一個探針分子上標記一個檢測的基團,所以,對長的克隆探針不適用。
此外,還可利用修飾的鹼基來增加雜交的穩定性和特異性。2-氫基腺嘌呤可替代寡核苷酸探針中的腺嘌呤通過形成3個氫鍵以增加雜交體的穩定性。另外,在G-C豐富的RNA探針中,可用次黃嘌呤代替鳥嘌呤以獲得特異的雜交。因為次黃嘌呤和鳥嘌呤間只形成2個氫鍵,有效地降低了雜交體的Tm值,這樣,Tm值與雜交溫度更接近,雜交的嚴格性就增加了,因此,也就增加了特異性。
很顯然,結合位點的不同和可檢測基團與檢測系統的不同,可派生出很多核酸探針標記方法。這是由核酸的化學結構和性質所決定的。只有在對核酸分子的探針-靶反應的化學本質有了深入了解之後,才能更好地理解後面內容。
概述
分子雜交基因探針根據標記方法不同可粗分為放射性探針和非放射性探針兩大類,根據探針的核酸性質不同又可分為DNA探針,RNA探針,cDNA探針,cRNA探針及寡核苷酸探針等幾類,DNA探針還有單鏈和雙鏈之分。下面分別介紹這幾種探針。
DNA探針
DNA探針是最常用的核酸探針,指長度在幾百鹼基對以上的雙鏈DNA或單鏈DNA探針。現已獲得DNA探針數量很多,有細菌、病毒、原蟲、真菌、動物和人類細胞DNA探針。這類探針多為某一基因的全部或部分序列,或某一非編碼序列。這些DNA片段須是特異的,如細菌的毒力因子基因探針和人類Alu探針。這些DNA探針的獲得有賴於分子克隆技術的發展和應用。以細菌為例,分子雜交技術用於細菌的分類和菌種鑒定比之G+C百分比值要準確的多,是細菌分類學的一個發展方向。加之分子雜交技術的高敏感性,分子雜交在臨床微生物診斷上具有廣闊的前景。細菌的基因組大小約5×106bp,約含3000個基因。各種細菌之間絕大部分DNA是相同的,要獲得某細菌特異的核酸探針,通常要採取建立細菌基因組DNA文庫的辦法,即將細菌DNA切成小片段後分別克隆得到包含基因組的全信息的克隆庫。
然後用多種其它菌種的DNA作探針來篩選,產生雜交信號的克隆被剔除,最後剩下的不與任何其它細菌雜交的克隆則可能含有該細菌特異性DNA片段。將此重組質粒標記後作探針進一步鑒定,亦可經DNA序列分析鑒定其基因來源和功能。因此要得到一種特異性DNA探針,常常是比較繁瑣的。探針DNA克隆的篩選也可採用血清學方法,所不同的是所建DNA文庫為可表達性,克隆菌落或噬斑經裂解后釋放出表達抗原,然後用來源細菌的多克隆抗血清篩選陽性克隆,所得到多個陽性克隆再經其它細菌的抗血清篩選,最後只與本細菌抗血清反應的表達克隆即含有此細菌的特異性基因片段,它所編碼的蛋白是該菌種所特有的。用這種表達文庫篩選得到的顯然只是特定基因探針。
對於基因探針的克隆尚有更快捷的途徑。這也是許多重要蛋白質的編碼基因的克隆方法。該方法的第一步是分離純化蛋白質,然後測定該蛋白的氨基或羥基末端的部分氨基酸序列,然後根據這一序列合成一套寡核苷酸探針。用此探針在DNA文庫中篩選,陽性克隆即是目標蛋白的編碼基因。值得一提的是真核細胞和原核細胞DNA組織有所不同。真核基因中含有非編碼的內含子序列,而原核則沒有。因此,真核基因組DNA探針用於檢測基因表達時雜交效率要明顯低於cDNA探針。DNA探針(包括cDNA探針)的主要優點有下面三點:①這類探針多克隆在質粒載體中,可以無限繁殖,取之不盡,製備方法簡便。②DNA探針不易降解(相對RNA而言),一般能有效抑制DNA酶活性。③DNA探針的標記方法較成熟,有多種方法可供選擇,如缺口平移,隨機引物法,PCR標記法等,能用於同位素和非同位素標記。
cDNA探針
cDNA探針
逆轉錄已成為一項重要的分子生物學技術,廣泛用於基因的克隆和表達。從逆轉錄病毒中提取的逆轉錄酶已商品化,最常用的有AMV逆轉錄酶。利用真核Mrna3’末端存在一段聚腺苷酸尾,可以合成一段寡聚胸苷酸(oligo(dT))用作引物,在逆轉錄酶催化下合成互補於mRNA的cRNA鏈,然後再用RNaseH將mRNA消化掉,再加入大腸桿菌的DNA聚合酶I催化合成另一條DNA鏈,即完成了從mRNA到雙鏈DNA的逆轉錄過程。所得到的雙鏈cDNA分子經S1核酸酶切平兩端後接一個有限制酶切點的接頭(linker),再經特定的限制酶消化產生粘性末端,即可與含互補末端的載體進行連接。常用的克隆載體是λ噬菌體DNA,如λgt,EMBL和Charon系列等。用這類載體可以得到包含104以上的轉化子的文庫,再經前面介紹的篩選方法篩選特定基因克隆。用這種技術獲得的DNA探針不含有內含子序列。因此尤其適用於基因表達的檢測。
RNA探針
RNA探針
近幾年體外轉錄技術不斷完善,已相繼建立了單向和雙向體外轉錄系統。該系統主要基於一類新型載體pSP和pGEM,這類載體在多克隆位點兩側分別帶有SP6啟動子和T7啟動子,在SP6RNA聚合酶或T7RNA聚合酶作用下可以進行RNA轉錄,如果在多克隆位點接頭中插入了外源DNA片段,則可以此DNA兩條鏈中的一條為模板轉錄生成RNA。這種體外轉錄反應效率很高,在1h內可合成近10μg的RNA產生,只要在底物中加入適量的放射性或生物素標記的NTP,則所合成的RNA可得到高效標記。該方法能有效地控制探針的長度並可提高標記物的利用率。
值得一提的是,通過改變外源基因的插入方向或選用不同的RNA聚合酶,可以控制RNA的轉錄方向,即以哪條DNA鏈以模板轉錄RNA。這種可以得到同義RNA探針(與mRNA同序列)和反義RNA探針(與mRNA互補),反義RNA又稱cRNA,除可用於反義核酸研究外,還可用於檢測mRNA的表達水平。在這種情況下,因為探針和靶序列均為單鏈,所以雜交的效率要比DNA-DNA雜交高几個數量級。RNA探針除可用於檢測DNA和mRNA外,還有一個重要用途,在研究基因表達時,常常需要觀察該基因的轉錄狀況。在原核表達系統中外源基因不僅進行正向轉錄,有時還存在反向轉錄(即生成反義RNA),這種現象往往是外源基因表達不高的重要原因。另外,在真核系統,某些基因也存在反向轉錄,產生反義RNA,參與自身表達的調控。在這些情況下,要準確測定正向和反向轉錄水平就不能用雙鏈DNA探針,而只能用RNA探針或單鏈DNA探針。
綜上所述,RNA探針和cRNA探針具有DNA探針所不能比擬的高雜交效率,但RNA探針也存在易於降解和標記方法複雜等缺點。
寡核酸探針
寡核酸探針
合成的寡核苷酸探針的優點
合成的寡核苷酸探針具有一些獨特的優點:
一。由於鏈短,其序列複雜度低,分子量小,所以和等量靶位點完全雜交的時間比克隆探針短,如20nt的寡核苷酸探針在濃度為100ng/ml,靶序列為1~100pg、1kb片段或3×10-18~3×10-16mol/L時,達到最大程度的雜交只需10min,而用2kb的克隆探針在同樣條件下達到完全雜交則需16h。
二。寡核苷酸探針可識別靶序列內1個鹼基的變化,因為短探針中鹼基的錯配能大幅度地降低雜交體的Tm值。
三。一次可大量合成寡核苷酸探針(1~10mg),使得這種探針價格低廉,與克隆探針一樣,寡核苷酸探針能夠用酶學或化學方法修飾以進行非放射性標記物的標記。儘管克隆探針較特異,但通過細心篩選序列和/或選擇相對長的序列(>30nt)亦可設計出非常特異的寡核苷酸探針。最常用的寡核苷酸探針有18~40個鹼基,合成儀可有效地合成至少50個鹼基的探針。
篩選寡核苷酸針的原則
下面是篩選寡核苷酸針的一些原則。
一。長18~50nt,較長探針雜交時間較長,合成量低;較短探針特異性會差些。
二。鹼基成分:G+C含量為40%~60%,超出此範圍則會增加非特異雜交。
三。探針分子內不應存在互補區,否則會出現抑制探針雜交的“髮夾”狀結構。
四。避免單一鹼基的重複出現(不能多於4個),如-CCCCC-。
五。一旦選定某一序更符合上述標準,最好將序列與核酸庫中核酸序列比較,探針序列應與含靶序列的核酸雜交,而與非靶區域的同源性不能超過70%或有連續8個或更多的鹼基的同源,否則,該探針不能用。
按上述原則選出的探針會增加成功的機會,選定後進行合成與標記,並摸索合適的雜交條件。方法是製備幾張點有特異靶DNA和不相關DNA的膜,各膜分別在不同溫度下與探針雜交,特異靶DNA雜交信號強而非特異DNA不產生任何雜交反應的就是最適雜交溫度。在進行點突變檢測雜交的反應時,洗膜條件和溫度物選擇往往更為重要。所選漂洗條件必需使野生型靶DNA與探針產生強的雜交信號而突變型靶DNA則不產生雜交信號,這可以通過逐漸提高洗膜溫度來完成。
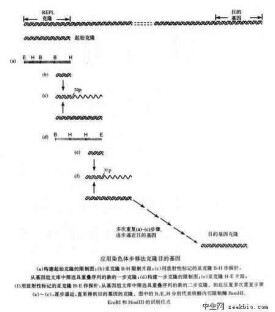
寡核酸探針
βADNA雜交體被切開后,5’端探針序列因只有8個鹼基,與雜交鏈結合不緊而解離,從而產生遊離的5’端標記8核苷酸單鏈。不被切開的βS雜交體尚可被另一個限制酶HinfI消化,該酶的識別位點緊靠DelI識別位點上游。βS雜交DNA經HinfI消化后,將釋出探針DNA的5’末端3核苷酸小片段。βADNA雜交體因已無HinfI識別序列,故而不能被HinfI消化。這樣βA和βSDNA經此寡核苷酸探針雜交和DelI及HinfI消化后,分別產生遊離的8核苷酸(8nt)和3核苷酸(3nt)片段,它們可以經電泳分離後進行放射自顯影而獲證實。藉此策略,可輕易將各種β珠蛋白突變型鑒別開,如純合野生型AA結果為僅有8nt片段,純合突變型SS則僅可檢出3nt片段,而雜合子AS型則兩種片段均存在。
基本信息
- 中文名
- 分子雜交
- 外文名
- molecular hybridization
- 拼音
- zá jiāo fēn zǐ
- 原理
- 氫鍵互補配對(關於DNA)