西比靈膠丸
由前庭功能紊亂引起的眩暈的對症治療葯
西比靈膠丸通用名稱是鹽酸氟桂利嗪膠囊(FlunarizineHydrochlorideCapsules),是藥品名稱,主要適用於典型(有先兆)或非典型(無先兆)偏頭痛的預防性治療或是由前庭功能紊亂引起的眩暈的對症治療。
份:鹽酸氟桂利嗪
化稱:()--[雙(-氟苯基)甲基]- -( - 丙烯基 – - 苯基) - 哌嗪二鹽酸鹽
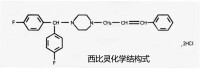
西比靈膠丸
分子式:CHFN·2HCl

西比靈膠囊

西比靈膠囊內含白色粉末實物圖
規格
5mg/粒(以氟桂利嗪計)x 20
偏頭痛的預防性治療
起始劑量:對於65歲以下患者開始治療時可給予每晚10mg,65歲以上患者每晚5mg。如果在治療中出現抑鬱、錐體外系反應和其它嚴重的不良反應,應及時停葯。如在治療2個月後未見明顯改善,則可視為病人對本品無反應,可停止用藥。
維持治療:如果療效滿意,患者需維持治療時,應減至每7天連續給葯5天(劑量同上)、停葯2天。即使預防性維持治療的療效顯著,且耐受性良好,在治療6個月後也應停葯觀察,只有在複發時才應重新服藥。
眩暈
每日劑量應與次相同同,但應在控制癥狀后及時停葯,初次療程通常少於2個月。如果治療慢性眩暈症1個月或突發性眩暈症2個月後癥狀未見任何改善,則應視為患者對本品無反應,應停葯。
不良反應
1、最常見的不良反應為:體重增加(11.3%),和/或食慾增加(4%)。
4、其它:口乾、多汗等。
禁忌
注意事項及
及個別病人在治療過程中乏力現象可能會逐漸加劇.此時應停止治療。
請在推薦劑劑量下使用。醫生應定期(特別是在維持治療期間)觀察患者,這樣可保證在出現錐體外系或抑鬱癥狀時能及時停葯。如果在維持治療時療效下降,亦應停止治療。
由於可能引起睏倦(尤其在服藥初期),駕駛車輛或操縱機器者應注意。
本品可能會引發錐體外系癥狀、抑鬱症和帕金森氏病,尤其是有此類病症發病傾向的患者如老年患者,所以此類患者應慎用。
孕婦及哺乳期婦女用藥
妊娠
哺乳
雖無本品隨人乳分泌的資料,但用哺乳期狗做的試驗表明鹽酸氟桂利嗪可隨乳汁分泌.其乳汁濃度較血中更高,故服用本品的婦女最好不哺乳。
兒童用藥
尚缺乏兒童用藥方面的資料。
老年用藥
老年患者慎用。
藥物相互作用
當本品與酒精、催眠葯或鎮靜葯合用時可出現過度鎮靜作用。
本品並不禁忌於使用β一受體阻斷劑的病人。
本品的葯代動力學不受托吡酯影響。在本品和托吡酯(50mg/12小時)合用治療期內,每隔12小時同服,觀察到偏頭痛患者體內氟桂利嗪的全身暴露量增加了16%;與單用本品的患者相比全身暴露量增加了14%。托吡酯的穩態葯代動力學也未受影響。
長期服用本品響會影響苯妥英,卡馬西平、丙戊酸鹽或苯巴比妥等藥物的分佈。使用此類抗癲癇藥物治療的癲癇患者體內本品的血葯濃度較給予相似劑量的健康受試者低。本品與卡馬西平、丙戊酸鹽或苯妥英合用時,卡馬西平、丙戊酸鹽或苯妥英的血漿蛋白結合率不受影響。
藥物過量
基於本品的藥理學特性,在過量服用時可能會出現鎮靜作用和虛弱,有報道個例超劑量服用的人(一次服用達600mg)出現嗜睡、激越和心動過速等癥狀。過量服用后1個小時內,可以進行洗胃治療。適當的情況下,也可以採用活性炭治療。尚無已知特定的解救藥。
鹽酸氟桂利嗪為選擇性鈣拮抗劑,可阻滯過量的鈣離子跨膜進入細胞內,防止細胞內鈣負荷過量,也可防止缺血缺氧時大量鈣進入神經元,改善腦微循環及神經元代謝,抑制腦血管痙攣、血小板凝聚及血液粘滯度增高等,此外還有細胞膜穩定作用。本品脂溶性高,易透過血腦屏障。
本品對心臟收縮和傳導無影響。
本品的非臨床中樞神經系統作用(如:鎮靜、唾液分泌和共濟失調)僅當暴露量遠遠超過人體的最大暴露量時才觀察到,與臨床使用的相關性很小。
對本品的安全性進行了一系列綜合的非臨床研究,包括:單次口服給葯的毒性(小鼠、成年及幼年大鼠、豚鼠)、腹膜內(小鼠和大鼠)、皮下(小鼠和大鼠)、靜脈(小鼠和大鼠)以及動脈內(大鼠)給葯;重複口服給葯的毒性(犬12個月和大鼠18個月多劑量的口服毒性)、靜脈毒性(犬3個月和大鼠1個月);口服給葯的生殖試驗測試大鼠的生育和總體繁殖能力;大鼠、兔和出生前、后大鼠的致畸性和胚胎毒性。對本品的致突變性進行了一系列廣泛的研究一包括:鼠傷寒沙門氏菌的體外位點和/或基因突變試驗、果蠅的伴性隱性致死試驗、人體內淋巴細胞的染色體畸變試驗、小鼠的體內微核和顯性致死試驗。利用口服給葯后小鼠和大鼠模型的壽命評估本品致癌性。
單次口服給葯的毒性試驗(小鼠的LD約為960-1896mg/kg;大鼠的LD約為343-1935mg/kg)結果與人體的最大治療劑量(對50kg的患者約為0.2mg/kg)相比,本品有很大的安全範圍。大鼠和犬重複口服給葯的毒性試驗結果顯示一些臨床作用可能與過度的藥理作用相關,但都是在遠遠超過藥物治療劑量時觀察到的(按mg/kg計算約為人體最大治療劑量的400倍),且與臨床使用的相關性很小。在生殖試驗中,本品對生育無影響且無致畸性。在非常高的劑量下(按mg/kg計算約為人體最大治療劑量的150-400倍),胎兒毒性小於母體毒性。本品沒有致突變性,也不是主要的致癌物。僅在小鼠體內達到毒性劑量水平時(按mg/kg計算約為人體最大治療劑量的50-100倍),才能觀察到由泌乳素介導的輕度乳腺增生和腫瘤生成。
在麻醉豚鼠的體內模型中,靜脈注射給予總劑量為9.87mg/kg(按mg/kg計算約為人體最大治療劑量的50倍)的氟桂利嗪,結果顯示對QTc間期和心電圖均無影響。
本品吸收良好,口服后2-4小時血葯濃度達到峰值,連續服用5-6周達到穩態。
吸收
氟桂利嗪經由胃腸道吸收良好(>80%),口服給葯后2-4小時內達到血葯濃度峰值。在胃酸降低的情況下(胃pH值升且),且生物利用度會適當下降。
分佈
氟桂利嗪與血漿蛋白結合率>99%。本品在健康人體和癲癇患者中均有很大的分佈容積,前者接近78L/kg,後者接近207L/kg,說明本品在血管外分佈廣泛。本品能迅速穿透血腦屏障,在腦中的濃度比在血槳中約高10倍。
代謝
氟桂利嗪在肝中代謝為至少15種代謝物。其主要代謝途徑為CYP2D6酶。
消除
氟桂利嗪主要以原形葯及其代謝物的形式經膽汁隨糞便排出。在給葯后的24-48小時內,約有3-5%的劑量以原形葯和代謝物的形式從糞便中排出,少於1%的劑量以原形葯由尿排出。其終末消除半衰期是高度可變的,在大多數個體中單次給葯后的變化範圍為5-15小時。一些受試者在一個延長期內(至第30天)可測到本品的血葯濃度(>0.5ng/mL),這可能是由於本品在其他組織中的重新分佈。
多劑量給葯
在每日一次的多劑量給葯下,氟桂利嗪的血葯濃度約8周后達到穩態,且比同劑量單次給葯的濃度高3倍。在5-30mg的範圍內,本品的血葯濃度與給藥劑量成正比。

基本信息
- 中文名
- 西比靈膠丸
- 分子量
- 477.42
- 分子式
- 活性成份
- 儲存
- 遮光,密封保存。
- 包裝
- 鋁塑板包裝,20粒/板/盒
- 有效期
- 36個月
- 執行標準
- 《中國藥典》2010年版二部
- 批准文號
- 國葯准字H10930003
- 生產企業