遺傳性耳聾
遺傳性耳聾
遺傳性聾指的是由於基因和染色體異常所致的耳聾。這種疾病是由父母的遺傳物質(包括染色體及位於其中的基因)發生了改變傳給後代而引起的耳聾,並且在於孫後代中以一定數量出現。在每1000個新生兒中就有一位患有先天性耳聾,其中60%以上是由遺傳因素引起的,遺傳性聾的群體發病率已超過27/1000,在所有耳聾病人中,遺傳性聾約佔50%。遺傳性聾分為綜合征性遺傳性聾及非綜合征性遺傳性聾兩大類。前者指除了耳聾以外,同時存在眼、骨、腎、皮膚等部位的病變,這類耳聾占遺傳性聾的30%;後者只出現耳聾的癥狀,在遺傳性聾中佔70%。
其中一部分病人,在出生后就對聲音沒有反應,如果不配戴助聽器和接受語言訓練,不但無法進行正常的交流,而且還阻礙了聾兒正常的智力發育,成為家庭和社會沉重的負擔;另一部分病人,只在10~30歲之間發病,表現為聽力下降,並隨著年齡增加不斷加重,以至達到極重度耳聾,這類病人雖然己學會說話,但由於聽力障礙,不能和別人進行正常的語言交流,也不能像正常人那樣看電影、電視,聽廣播、音樂,對本人和家庭都是個極大的痛苦。
隨著現代科學技術的進步以及分子遺傳學的飛速發展,人們對遺傳性聾的認識不斷加深,相信在不久的將來,遺傳性聾這一醫學難題必將被攻克,造福於聾病患者。
在此提醒讀者的是,家庭中關有兩位以上的聾人,應儘早到醫院進行檢室和諮詢,以儘早排除遺傳性聾,並盡量避免在下一代出現聾病患者。
遺傳性耳聾的分類
遺傳性耳聾的分類方法目前尚未統一,在此,僅就遺傳方式,病變位置,發病時間和伴發疾病等進行闡述。
⑴常染色體隱性遺傳性聾是遺傳基因位於常染色體上、由隱性基因控制的遺傳。此類耳聾只有在兩個分別來自父母的等位基因均為致聾基因時才出現耳聾。隱性遺傳性聾到目前為止占單基因突變的80%.儘管大多不發病,但基因攜帶者將把相同基因型傳遞給他們25%的子女。含有耳聾隱性基因的嬰兒如果是家庭中的第一位發病者,由於無相關的全身其他異常及無耳聾家族史,耳聾可在不被察知的情況下出現。
⑵常染色體顯性遺侍性聾指遺傳基因位於常染色體上,並由顯性基因控制的遺傳。此類耳聾占基因性聾的10%-20%,嬰兒接受來自父母之一方的致病基因即可發病。耳聾患兒既可以是患病父母基因表達的結果,也可以是父母新的基因突變發生所致。基因攜帶者幾乎總是患者,但患者之間臨床表現程度有較大差異。
⑶ 性連鎖遺傳致聾基因位於X染色體上,隨X染色體傳遞。此類耳聾占基因性聾的1%-2%,包括隱性遺傳和顯性遺傳。女性在其中的一條x染色體含有耳聾隱性基因者聽力正常,雜合子男性將它傳給所有兒女。若為隱性伴性遺傳,一半男性受累.而女性必須純合子才受累,未受累女性成為這種遺傳性狀的攜帶者。顯性遺傳者,若母親耳聾,子女中約有1/2人發病;如父親患病,則全部女兒均患病。
⑷.線粒體基因突變發病率較低,呈母系遺傳。
病變位於外耳和(或)中耳,引起傳導性耳聾,如外耳道狹窄或閉鎖、聽小骨畸形、耳硬化症等。
病變位於內耳,引起感音神經性耳聾。
病變累及外耳和(或)中耳和內耳者,則引起混合性耳聾,此型比較少見。
先天性遺傳性耳聾:耳聾於出生后即已發生,且出生后不再發展的遺傳性聾屬先天遺傳性聾。
遺傳性進行性耳聾:出生時耳的各部分均正常發育,聽力正常,而於出生后某一年齡階段方始出現進行性聽力下降,最後發展為嚴重的耳聾。
⑴單純型遺傳性聾:耳聾為發病個體唯一的遺傳性疾病,其它器官無遺傳性損害。
⑵伴有其它器官遺傳疾病的耳聾綜合征:患者除遺傳性聾外,尚伴有身體其他器官的遺傳性疾病,如眼、骨骼系統、神經系統、腎臟、皮膚、內分泌系統、代謝性疾病等。
根據迷路的解剖特徵,先天性聾包括遺傳性聾和其他出生前內耳畸形已定型的非遺傳性聾,可分成4種基本類型。
(發育不全型):這是最嚴重的內耳畸形,其特徵是部分或整個迷路不發育(包括耳蝸和前庭),偶可見殘餘膜迷路結構。蝸神經及前庭神經可存在或缺如,一般無聽覺。
(骨及膜迷路的各種畸形):為常染色體顯性遺傳,耳蝸前庭發育不全,耳蝸可能部分發育,通常只有基底部的1周半或2周,球囊、橢圓囊及半規管可呈發育畸形,蝸神經及前庭神經其神經節通常存在或部分存在。患者可有殘餘聽力,但很少有可用的言語聽力。
(膜迷路畸形型):骨迷路和膜性橢圓囊及半規管發育完善,但膜性蝸管及球囊則發育不全,蝸管萎縮。螺旋器和血管紋呈未發育的索狀結締組織,球囊壁坍陷於發育不全的感覺上皮和耳石膜上。本型是遺傳性先天性聾中最常見者。患者可有部分聽力。
(中度膜迷路畸形型):蝸管發育不全,以基底周的螺旋養及其鄰近的神經節受患最深,造成高頻聽力損失。因其低頻聽力尚存,配戴助聽器可有幫助。
較多見,包括單耳和雙耳聾。相對於遺傳異質性的程度來說,經過臨床檢查僅能根據病變部位將耳聾分為傳導性、感音神經性和混合性耳聾3種。傳導性耳聾如由外耳道閉鎖所引起者,其根據病變程度可分3組,第一組外耳道發育不全,小鼓膜,中耳腔正常或偏小;第二組外耳道閉鎖,中耳腔小而畸形;第三組為第二組的特徵加上中耳腔明顯發育不全,或無中耳腔。感音神經性耳聾可為單側或雙側性,嚴重程度可為輕度到重度不等。對於出生時已耳聾者,其耳蝸病變一般已穩定,故屬非進行性。而進行性者出生時耳蝸發育正常,於生后某一年齡方始出現退性性變。由於起病時言語發育多已完成,故患者一般聾而不啞。
在胚胎髮育中,皮膚、毛髮、指(趾)甲、部分色素、內耳及中樞神經系統均發源於外胚層。因此,耳聾可伴隨以上器官的異常。①Wamdenburg綜合征(先天性耳聾眼病白額發綜合征):是先天性耳聾中較常見的一種,占所有先天性耳聾的2%-5%,主要為常染色體顯性遺傳,幾乎100%外顯,但表達程度各異。臨床特徵為前額白髮,鼻根增寬,內眥外移,虹膜異色及聾啞,聽力損害可出現於20%—30%的患者,單側或雙側,程度不一。根據內眥是否外移分為兩型,內眥外移為I型,否則為Ⅱ型。②伴隨耳蝸性聾的其他皮膚病變:如白化病,色素異常,先天性非變應性濕疹,角質厚皮病,白甲病,指(趾)甲營養不良及錐形牙等。
①Usher綜合征(聾啞、網膜色素變性綜合征):屬常染色體隱性遺傳性疾病,其特點為聾啞伴視網膜色素變性,共分3型:I型表現為重度耳聾、前庭反應缺如,夜盲出現於嬰兒期或青少年期,成年早期發生法定盲。Ⅱ型表現為先天性感音神經性耳聾,聽力曲線呈下斜型,前庭功能正常,夜盲出現於青少年期,成年早期或中年出現法定盲。Ⅲ型特點為出生時聽力可正常或中度聽力障礙,但以後可發展,聽力曲線呈下斜型,前庭功能減退並可發展,夜盲的程度和進展可不等。②Hallgren綜合征:耳聾伴前庭功能異常;有非典型性進行性視網膜色素變性,以致視野縮小及夜盲;有多發性神經炎性疾病如小腦性共濟失調、眼源性眼球震顫及肌萎縮等。③Refsum綜合征(遺傳性運動失調—多發性神經炎綜合征):其特點是耳聾、視網膜色素變性、多發性神經炎及銀屑病等。④Cockayne綜合征(網膜萎縮侏儒耳聾綜合征):較少見,特點為耳聾、視網膜萎縮、智力發育遲鈍及侏儒病等。⑤Laurence-Moon-Bandet-Biedl綜合征(肥胖視網膜色素變性—多指(趾)畸形):少見,特點是耳聾、智力發育遲鈍、肥胖症、多指(趾)畸形及性功能減退。
較罕見。①Hürler綜合征(怪面形耳聾綜合征、糖胺聚糖沉積病IH型):感音性耳聾伴骨骼變形、矮小、失明、肝脾腫大。耳聾始於嬰兒期,逐漸加重,輕度至中度耳聾,有時可伴進行性智力衰退及角膜混濁。②Richards—Rundle綜合征:耳聾伴軀幹共濟失調,智力衰退,性腺功能減退。患兒到5—6歲時,聽力完全喪失。
此類耳聾多屬傳音性,但如病變涉及內耳則同時亦可有耳蝸性聾。
①Treacher Collins綜合征(頜面骨發育不全及耳聾綜合征):為常染色體顯性遺傳,呈不同程度的外顯。近60%的病例可由新突變的基因所引起,故而臨床上可呈散髮狀。其特徵為:a.顱骨狀況為眶上緣發育差,顴骨發育差,甚至缺如但雙側對稱,乳突氣化差或明顯硬化,鼻竇小或完全不發育,兩眶距過寬,下頜骨踝狀突嚴重發育不全。b.眼部表現為瞼裂短、外方下斜,下瞼外1/3通常缺如,約半數患者睫毛少或缺如,下淚點及瞼板腺可缺如。c.耳部畸形可表現為耳郭杯狀、下斜或低位至下頜骨角;多數患者外耳道狹小且扭曲,約1/3的患者外耳道可缺如;多數病例中耳腔小甚至為結締組織所充填;聽小骨畸形,前庭耳蝸畸形或部分缺如;乳突發育差甚至氣房缺如。d.鼻部因顴弓和眶骨發育不全而顯突出,鼻孔狹小,鼻翼軟骨發育差,也有后鼻孔閉鎖的報道。e.口部表現為不同程度的齶裂及齶咽畸形.巨口(頰橫裂)可為單側或雙側。f.智力發育通常正常,但也有發育遲緩的報道。
②Goldenhar綜合征:又稱眼耳椎骨發育異常、Goldenhar-Gorlin綜合征、第一鰓弓綜合征、第一和第二鰓弓綜合征、牛面發育異常等。臨床特徵為各種程度的面部不對稱;患側可有瞼裂狹小;各種程度的耳郭畸形,外耳道狹窄或閉鎖,可有面神經徑路異常和顱底畸形;5%—15%的病例有智力發育障礙;一側腎臟缺如、雙輸尿管、腎異位、腎血管異常、腎盂積水;患側面部前後徑及椎骨體積變小,顳頜關節前下移位,眼眶縮小,頸椎融合;可見單側頰橫裂或假性頰橫裂;另也有心臟、肺及胃腸道畸形的報道。
③Klippel-Feil綜合征(頸—跟—聽覺綜合征):其特點為骨骼發育異常,包括先天性短頸、頸椎融合、頭偏斜、脊柱側凸及脊柱裂等,約1/4—1/2患兒伴有感音性和(或)傳導性耳聾。耳聾可發生於出生時,也可延遲起病。在延遲起病者,耳聾的程度、聽覺減退的發展狀況可差別很大。
④Willebrand綜合征(頸眼耳綜合怔):特點為頸短而強直(因頸椎融合),髮際低,雙側展神經癱瘓及先天性感音性聾。⑤Madelung綜合征:特徵為身材矮小、前臂短、下肢短,有腕骨、脛骨、腓骨的多發性畸形。患者多為女性,男女發病率之比為1:4。多屬聽骨鏈異常所致傳導性耳聾,亦可以感音性聾為主。
①Pendred綜合征(先天性甲狀腺腫耳聾綜合征):為常染色體隱性遺傳,患兒出生時即有耳聾,嚴重者成為聾啞,也可為遲發性先天性進行性聾。甲狀腺腫多在少年兒童期出現,亦有在青春期后才顯著,為碘代謝異常所致。甲狀腺功能正常和低下者各佔50%。②DiGeorge綜合征(胸腺發育不良、Ⅲ、Ⅳ咽囊綜合征):其特點為第3、第4咽囊及中耳、內耳畸形,先天性胸腺及甲狀旁腺缺損。③Jervell—Lange—Nielsen綜合征(耳聾、心電圖異常綜合征):為常染色體隱性遺傳,特點為先天雙側嚴重耳聾,心電圖Q—T間期延長,反覆發作暈厥,甚至猝死。
⑶染色體異常:Down綜合征(21—三體綜合征、先天愚型),是最常見的染色體異常,患者常有傳導性耳聾;E18—三體綜合征是指額外的染色體位於E組的第18對中。臨床表現為低位耳、耳部畸形、小頜、示指(食指)彎曲在中指之上,枕骨凸出,患者通常在出生不久即死亡;D13-15—三體綜合征,指額外的染色體在D組的第13或14或15對中.表現為低位耳.外耳、中耳、內耳畸形,唇裂、齶裂,小眼,隱睾及心臟病,患兒常在出生后不久即夭折。
1)家族性進行性感音神經性聾:為發育完善的耳蝸發生變性所致。耳聾常為雙側性,一般於兒童期(8-11歲)或成年早期開始,並逐漸加重。顳骨切片可示耳蝸基底周螺旋器和螺旋神經節變性或消失,以及血管紋的不規則變性,聽力曲線常示高頻聽力減退,或呈平坦形,或盆形。
詳見有關章節。
⑴伴隨其他異常綜合征
1)Branchio-oto-renal綜合征(腮裂、耳、腎綜合征、BOR綜合征):為常染色體顯性遺傳
病,不同程度的外顯。臨床表現為鰓裂瘺管或囊腫,耳郭畸形,聽力損害及不同程度的腎臟畸形。
(家族遺傳性出血性腎炎耳聾綜合征):為常染色體顯性或性連鎖顯性
遺傳病,臨床特徵為腎小球腎炎及感音神經性耳聾。患者如有以下4個標準中的3個,即可珍斷為此綜合征。①血尿家族史,或伴有腎功能衰竭。②腎臟活檢電鏡檢查示腎小球腎炎。③眼部徵象,如前錐形晶體、黃斑周圍微粒、白內障和眼性跟震等。④兒童期出現高頻聽力減遲,並呈進行性發展。
為顱、干骺端發育不良,聽力障礙多呈傳導性聾,偶為耳蝸性聾,伴股骨豁開,顱骨變化類似骨硬化病,可因顱骨孔狹窄偶爾猝發腦神經癱瘓。
如Hurler、Klippel-Feil、Refsum、Richards-Rundle等綜合征都可有延遲性耳聾發生。
人類精卵細胞核的染色體是遺傳物質的載體。精卵細胞中共有23對46條染色體。其中22對44條稱常染色體,另外一對稱性染色體。性染色體決定性別,男性為XY,女性為XX。人類遺傳物質的特性是能夠準確地複製自己,在遺傳的傳遞中,引用了“基因”這個概念。基因是比染色體更小的單位,不能在顯微鏡下觀察到,只能通過家譜分析來判斷基因的存在。先天性耳聾的遺傳類型有以下3種:
指親代帶有的病理性基因是隱性的,只有在基因成對時(純合子),疾病才能表現出來,這種方式稱為常染色體的隱性遺傳,是先天性耳聾的主要遺傳方式。
指親代帶有的病理性基因是顯性的。只要有一個基因傳給子代,子代就會複製親代疾病,即個體只要有一個顯性致病基因就會發病。如果雙親之一為先天性遺傳聾,其子女發生耳聾危險率為50%。
指致聾基因不在常染色體上而是在性染色體上,也分為顯性遺傳和隱性遺傳。若遺傳基因結合在X染色體上,伴性隱性遺傳聾者,其子女1/2為耳聾,另1/2為致聾基因攜帶者;伴性顯性遺傳聾者,其子女男性全部為耳聾患者,女性1/2為耳聾患者。
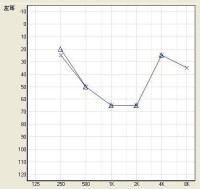
盆狀聽力圖多見於遺傳性聽力損失
聽神經再生還原療法
2.具有行氣開竅,改善內耳供血、增強耳內代謝,提高毛細胞興奮性等,打通血液循環障礙,營養修復再生耳細胞,激活耳蝸神經,使耳部細胞得以修復再生。
3.具有活血化瘀、清除自由基、修復病變細胞、解除耳沉痛,短期即可促進變性、萎縮、壞死的聽覺神經細胞修復再生,經全面規範治療,短期內可消除耳鳴,恢復正常聽力。
4.局部光波治療的方法,可調節血管功能,使血流加速,改善內耳血液淋巴循環;加強組織代謝,糾正內耳缺氧狀況並能及時排出有害物質,促進耳蝸功能及聽功能的改善與恢復。
5.電針治療耳聾、耳鳴是脈衝電磁場直接介入患者中耳及內耳,電波能改善局部血液循環,改善組織通透性,改善耳蝸供血,有利於恢復耳蝸的正常生理功能。
基本信息
- 中文名
- 遺傳性耳聾