哈米特方程
哈米特方程
哈米特方程(Hammett方程)是一個描述反應速率及平衡常數和反應物取代基類型之間線性自由能關係的方程。
所研究的反應物是苯甲酸以及間位和對位取代的苯甲酸。通過測定這些苯甲酸衍生物的pKa值,可以獲得它們的取代基常數(σ),也就是對取代基電子效應的量度。在其他反應中,通過將反應的log(k/k0)對σ作圖,所得圖像的斜率(ρ)可以表示反應受取代基影響的靈敏度,從而提供了速率控制步驟中反應中心的電荷變化的信息。這個方程的基本概念是,對於取代基類型不同但其他結構相同的反應物,反應的活化自由能與吉布斯自由能的差成一定的比例關係。
對於取代基類型不同但其他結構相同的反應物,反應的活化自由能與吉布斯自由能地差成一定的比例關係。路易斯·普拉克·哈米特於1935年提出此概念,並於1937年首先提出方程的形式。
在芳香族化合物,尤其是取代苯的反應中,k0是不含取代基反應物的反應速率常數,k是含有取代基的反應物的速率常數。它們之間存在如下關係:σ稱為“取代基常數”,只與取代基的類型有關;ρ稱為“反應常數”,只與反應的類型有關,與取代基類型無關。哈米特方程的另一種形式是描述反應中平衡常數的關係,即:
K0是不含取代基反應物的反應平衡常數,K是含有取代基的反應物的反應平衡常數。
在芳香族化合物,特別是苯類化合物的反應中,若不含取代基的反應物的反應速率常數為k0,當引入取代基后,其反應速率常數為k,則k與k0之間有如下關係:
lgk=lgk0+σρ
這就是哈米特方程。式中σ為取代常數,其值決定於取代基團的性質和位置(如鄰位、間位、對位);ρ為常數,決定於反應類型。
哈米特方程的另一種形式是描述反應平衡常數的關係,即:
lgK=lgK0+σρ
式中K0為不含取代基的反應物的平衡常數;K為含取代基的反應物的平衡常數。
規定氫的σ值為0.00,苯甲酸和取代苯甲酸的水溶液在25℃時電離反應的ρ為1。根據苯甲酸的電離常數K和取代苯甲酸的電離常數K,即可求得各種取代基的相對σ值:
表1列出了一部分基團的σ值。給電子基團的σ為負值,吸電子基團的σ為正值。由σ數值的大小,可定量表示取代基給電子性或吸電子性的強弱。σ是各種效應的綜合結果。
根據已求得的σ值,可用哈米特方程求得各反應的ρ值。表2列出了部分反應的ρ值。ρ值可為<0、0、0<ρ<1、≥1等各種可能的數值。人們認為ρ的數值與反應過渡態的荷電情況有關。當過渡態與反應物相比具有正電性時,則ρ0;電性無變化時,則ρ=0。如果與ρ=1的標準反應相比,則ρ1時,則過渡態的電荷改變比標準反應具有更強的負電性,ρ值越大,則負電性越強。測定反應的ρ值,已成為研究有機反應機理的有效手段之一。
哈米特方程分子式
氫為取代基時的取代基常數和反應常數被定為1(即苯甲酸自身),其他取代基的取代基常數是在此基礎上,通過以下公式計算獲得的:
| Hammett取代基常數 | ||
| 取代基 | 對位常數 | 間位常數 |
| –NH2 | -0.66 | -0.161 |
| –OCH3 | -0.268 | +0.115 |
| –OC2H5 | -0.25 | +0.15 |
| –N(CH3)2 | -0.205 | -0.211 |
| –CH3 | -0.170 | -0.069 |
| –H | ||
| –F | +0.062 | +0.337 |
| –Cl | +0.227 | +0.373 |
| –Br | +0.232 | +0.393 |
| –I | +0.276 | +0.353 |
| –NO2 | +0.778 | +0.710 |
| –CN | +1.000 | +0.678 |
| 參考資料:Hammett1937 |
右表中列出了一些比較常見的取代基常數值。可以看出,取代基常數值是取代基電子效應的量度,其效果是誘導效應(±I)和共軛效應(±M)的綜合效果。σ值最大的取代基是氰(qing2)基和硝基,是典型的吸電子基團,相應的芳香羧酸由於陰離子更加穩定,使羧酸酸性更強,pKa更小。
接下來是各種鹵素取代基。它們有吸電子的誘導效應和供電子的共軛效應,但總體上仍是吸電子的,故σ仍為正值。對位取代的鹵素由於可以和苯環共軛(e4),產生下面的共振式結構,鹵素的電子通過苯環向羧基轉移,使得質子不易離去,酸性較低。間位取代的鹵素由於不存在這個現象,因此σ值大於對位的σ值。
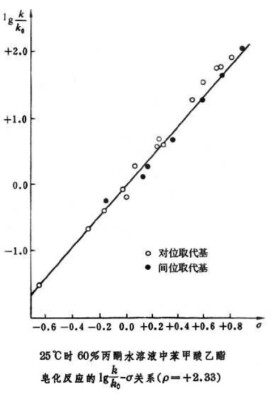
在取得各種取代基常數值后,哈米特又用不同取代的芳香羧酸發生某一特定反應,並計算出這些反應的速率常數。他發現,如果用log(kX/kH)對σ作圖(Hammett圖),可以發現兩者符合很好的線性關係。log(kX/kH)-σ圖的斜率記作ρ,也就是此類型反應的反應常數。苯甲酸解離的反應常數定為1。
大部分反應都存在這種線性關係,其中一個比較典型的反應是30°C時,取代苯甲酸乙酯在水/乙醇混合物中發生水解的反應。這個反應的反應常數值為+2.498。
其他很多反應的ρ值都是已知的。以下是哈米特在原始文獻中列出的一些反應常數值:
取代肉桂酸酯在乙醇/水混合物中的水解反應(+1.267);取代苯酚在水中的解離反應(+2.008);酸催化下取代苯甲酸在乙醇中的酯化反應(-0.085);酸催化下取代苯乙酮在乙醇/水/鹽酸混合物中的溴化反應(+0.417);69.8°C時,取代氯化苄在丙酮/水混合物中的水解反應(-1.875)。由於ρ是由速率商的對數對取代基常數作圖得到的,因此一個反應的ρ值可以看作是,相對於苯甲酸解離反應而言,這個反應的反應速率受取代基變化的靈敏度的定量測量。如果吸電子基團對反應起加速作用,很明顯ρ>0,哈米特圖的斜率是正值;如果給電子基團對反應起加速作用,ρ<0,哈米特圖向後傾斜。也就是說:
ρ>1,反應速率比苯甲酸解離的反應更容易受取代基影響,且速率控制步驟是負電荷累積的過程;0<ρ<1,反應速率比苯甲酸解離的反應更不容易受取代基影響,且速率控制步驟是負電荷累積的過程;ρ=0,反應速率不受取代基影響,且速率控制步驟中反應中心電荷無明顯變化;ρ<0,速率控制步驟是反應中心正電荷累積的過程。這是哈米特方程用於測定有機反應機理的基礎。當一個有機反應存在兩個未經證實的機理時,可以通過用不同取代的芳香族化合物原料分別發生反應,計算出反應速率和ρ值,並判斷出速率控制步驟時反應中心是否有電荷的明顯變化。如果ρ在-1與1之間,則基本上可以否定數控不涉及正/負電荷累積的機理。有趣的是,協同反應(如DA反應)由於基本不涉及電荷變化,故其ρ值大多在-1與1之間,且ρ值的正負可用於判斷哪個反應物分子出HOMO,哪一個出LUMO。
基本信息
- 中文名
- 哈米特方程
- 外文名
- Hammett equation
- 提出者
- L.P.哈米特
- 提出時間
- 1937年