羥醛反應
羥醛反應
羥醛反應(英語:Aldol reaction)是有機化學中形成碳-碳鍵的重要反應之一。它是指:具有α氫原子的醛或酮在一定條件下形成烯醇負離子,再與另一分子羰基化合物發生加成反應,並形成β-羥基羰基化合物的一類有機化學反應。該反應由查爾斯·阿道夫·武茲和亞歷山大·波菲里耶維奇·鮑羅丁於1872年分別獨立發現,反應連接了兩個羰基化合物(最初反應使用醛)來合成新的β-羥基酮化合物。此類產物稱作“羥醛”(Aldol),取自醇羥基的“羥”(ol)字和醛類化合物的“醛”(ald)字形成的名稱。
一個典型的現代羥醛加成反應即酮的烯醇負離子對醛的親核加成。一旦反應發生,羥醛產物有時可以失一分子水形成α,β-不飽和羰基化合物,這就稱作:羥醛縮合反應。在羥醛反應中可以使用各種親核試劑,包括烯醇、烯醇負離子、酮的烯醇醚、醛和其他羰基化合物。與之反應的親電試劑通常是醛或酮(這裡有許多變化如曼尼希反應)。若親核試劑和親電試劑不同,反應稱作:交叉羥醛反應;若親核試劑和親電試劑相同則稱作:羥醛二聚化反應。
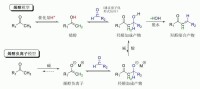
典型的羥基縮合反應
羥醛反應第一個具里程碑意義的事件出現於1957年,當時美國西北大學的H.E.齊默曼(Zimmerman)和M.D.特拉克斯勒(Traxler)為解釋格氏試劑介導的Ivanov反應中反式產物佔優勢的問題,提出了著名的六元環過渡態模型,後人常稱之齊默曼-特拉克斯勒模型。該模型首次從立體化學角度對羥醛反應進行剖析,指出烯醇鹽構型與產物立體化學之間的對應關係,成為羥醛反應歷史上的第一個理論突破並在很長一段時間內成為後續研究的唯一指導性理論。以後眾多的實驗結果也證明這個模型非常成功,根據模型做出的實驗設計大多得到了預期的立體化學結果。
二十世紀六十年代,核磁共振技術在為立體化學的發展形成了強大的推動力,它為羥醛反應研究帶來了極大便利。通過氫譜的積分、化學位移、偶合常數等數據能方便、快捷地實現產物立體化學的分析鑒定。在此背景下羥醛反應的研究開始逐步升溫。
1973年日本北里大學教授向山光昭發明了羥醛反應中使用硅醚形式來穩定烯醇的方法,該發現為羥醛反應研究揭開了新的一頁。向山羥醛反應通過事先製備烯醇硅醚與醛酮混合后在路易斯酸催化下得到羥醛產物。由於產物的立體化學與烯醇硅醚的構型的對應關係不再符合齊默曼-特拉克斯勒模型,其又提出向山開鏈過渡態理論。通過路易斯酸在向山羥醛反應中的研究,發展了羥醛反應立體化學控制技術,並成為該反應立體化學三大控制方向之一。
二十世紀七十年代到八十年代是有機化學發展史上的重要時期,當今眾多不對稱合成技術就是在這十多年間得以發展的。羥醛反應立體控制技術在這段期間發生了重大的突破。日本的向山光昭、正宗哲,美國的大衛·埃文斯、希思科克、A.E.梅耶等都在這段時間對羥醛反應立體控制技術做出了重大的貢獻。1981年哈佛大學教授大衛·埃文斯發明了手性惡唑烷酮配體介導的不對稱羥醛反應技術,第一次實現了高對映選擇性的不對稱羥醛反應。
羥醛結構單元存在於許多分子(包括天然產物和合成分子)中。例如,通過羥醛反應大規模合成的日用化學品季戊四醇及心臟病藥物阿托伐他汀。羥醛反應之所以應用廣泛是因為它將兩個相對簡單的分子結合成一個較複雜的分子,通過形成兩個新的手性中心(於羥醛產物的α-碳原子上,在下述分子式當中標註)增加了分子複雜性。現代化學方法學不僅可以做到羥醛反應的高收率,而且能夠控制反應產物的相對和絕對立體化學構型。這種選擇性合成特定的立體異構體非常重要,因為不同的立體化學異構體可能具有完全不同的化學或生物特性。
如手性羥醛單元在聚酮化合物中較常見,聚酮是一種在生物有機體中發現的分子。在大自然中,聚酮通過酶進行多重克萊森縮合反應。這些反應產生的1,3-二羰基化合物可衍生出各式各樣有趣的結構,其中一些具有強效的生物活性,如強效免疫抑製劑他克莫司、抗癌藥物盤皮海綿內酯及抗真菌藥物兩性黴素B。儘管這些化合物中許多曾經認為幾乎不可能被合成,但羥醛反應方法學使之變為可能,且在許多合成中成為關鍵步驟。
羥醛反應可基於兩種不同的機理進行:
羰基化合物如醛或酮可轉化為烯醇或烯醇醚。它們在碳原子上都具有親核性,可以進攻一些活潑的質子化羰基化合物,如質子化的醛,稱為“烯醇機理”。
羰基化合物或碳原子上含活潑氫的有機物,可於羰基位去質子化形成烯醇負離子,而該離子形態比烯醇和烯醇醚更具親核性,可直接進攻親電試劑。常見的親電試劑為醛類化合物,而酮的活性相對較低,這類反應機理稱為“烯醇負離子機理”。
若反應條件特別劇烈,如:甲醇鈉作鹼,甲醇為溶劑的迴流條件下,就會發生縮合反應;而這種情況可以通過低溫條件並使用溫和的鹼加以避免,如:二異丙基氨基鋰作鹼,四氫呋喃作為溶劑,在下反應。雖然羥醛加成反應通常能進行到底,卻並非不可逆。用強鹼來處理羥醛加成產物,可導致逆向-羥醛裂解而得到起始原料。羥醛縮合通常認為是不可逆反應但交叉羥醛反應動力學研究表明其實際上是可逆反應。
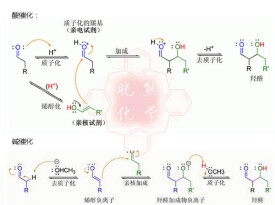
在酸催化條件下,反應機理的起始步驟是羰基在酸-催化下異構化為烯醇。酸還通過質子化活化另一分子羰基,使其具有高度親電性。烯醇在碳原子上具親核性,能進攻質子化的羰基化合物,而後去質子化形成羥醛。通常最後還會繼續脫水得到不飽和羰基化合物。以下展示了典型的酸-催化下的醛自身縮合反應:
酸催化的羥醛反應機理
若催化劑是溫和的鹼,如氫氧根離子或醇負離子,則羥醛反應可通過形成共振-穩定的烯醇負離子而親核進攻另一分子羰基化合物。其產物為羥醛產物的醇鹽,而後自身脫水得到不飽和羰基化合物。反應式展示了鹼-催化下的醛自身進行羥醛反應的機理:
鹼催化的羥醛反應,圖例:
鹼催化的脫水反應,此反應通常被錯寫為簡單一步,,
雖然某些情況下反應僅需要催化量的鹼,但大多數的反應歷程都需要等當量的強鹼,如二異丙基氨基鋰(LDA)或六甲基二硅氮基鈉()。在這種情況下,烯醇負離子的形成是不可逆的,直到酸化后處理中羥醛產物的金屬醇鹽被質子化,羥醛產物才得以形成。
有機反應過渡態是控制反應的關鍵要素之一。羥醛反應的第一個過渡態模型是1957年由霍華德·齊默曼和馬喬里·特拉克斯勒提出的六元環過渡態模型即如下圖所示齊默曼-特拉克斯勒模型,M是由鹼帶入的金屬離子。運用建立在環己烷上的構象分析理論可以看出,醛分子在受烯醇進攻時的構象形態主要是使羰基兩側較大的取代基處於六元環平伏鍵的位置,因為可以使各官能團之間空間位阻最小,過渡態的能量也最小,而導致優先形成相應立體構型的產物.
從圖中同樣可以看出,碳上的立體構型由烯醇鹽的構型決定。Z型烯醇主要生成順式羥醛,而E型烯醇生成以反式羥醛為主的產物。值得注意的是,上面的順反構型是相對構型,因為存在兩種順式產物和兩種反式產物。換言之,上面每個產物對應兩種可能的絕對構型。
羥醛加成的反應“控制”問題最好通過一個實例來說明。請考慮以下假定反應的結果:在此反應中,兩個不對稱酮使用乙醇鈉進行縮合反應。乙醇鈉的鹼性不能將其中任意一個酮完全去質子化,但可以將兩種酮都形成少量的烯醇鈉鹽。這意味著兩種酮分子不僅都是潛在的親電試劑,而且都可以通過形成烯醇負離子成為親核試劑。最後的結果是兩種親電試劑和兩種親核試劑形成的四種產物:
所以,若希望得到交叉-產物中的一種就必須控制其中一個成為親核試劑,另一個保持親電的羰基形式。
最簡便的控制方法是兩種參與反應的羰基分子中只有一種具有氫原子,而只有這種分子才能異構化成為烯醇式。例如,丙二酸酯和苯甲醛反應則只會形成一種產物。因為只有丙二酸酯具有氫原子而成為親核試劑,不能進行烯醇化的苯甲醛則只能成為親電試劑:
丙二酸酯非常易於去質子化,這是因為它的羰基位和另外一個羰基連接。1,3-二羰基形式能讓形成的烯醇負離子更穩定,所以不需要很強的鹼就能讓其烯醇化。此效應可以引申出另外一種情況:即使羰基旁有兩種氫原子,卻仍然可以控制形成哪種烯醇式。若兩種氫原子酸性具有較大差異,就會導致其中一個較強酸性質子被鹼中和形成烯醇,而另一個酸性較弱氫原子則不被鹼影響。這種類型的控制,只存在於酸性區別足夠大而鹼不過量的情況下。其應用在亞甲基臨近於兩個羰基或者氰基的分子中較常見,如Knoevenagel縮合反應和丙二酸酯合成的第一步。
常用的解決方案是讓其中一個羰基化合物先形成烯醇負離子,然後在動力學控制下加入另一個羰基化合物。動力學控制意味著正向羥醛加成反應必須明顯快於反向羥醛反應。要達成這個目的必須滿足其他兩個條件:1、量化形成其中一種烯醇負離子;2、正向羥醛反應明顯快於烯醇離子的交換過程。常見的動力學控制操作是使用二異丙基氨基鋰在下和酮反應得到烯醇負離子,而後緩慢加入醛。
烯醇是醛或酮的異構體。在存在少量酸或鹼的條件下烯醇和醛酮可以相互轉化,稱為互變異構,或烯醇和醛酮互為互變異構體。酸性或中性條件下烯醇不穩定,互變異構的反應平衡有利於醛酮而不利於烯醇,所以醛酮中只含有微量的烯醇異構體。如下圖所示:
烯醇負離子可通過兩種方法獲得:1、強鹼(“硬條件”)2、路易斯酸和弱鹼(“軟條件”)。
要進行去質子化,立體電化學需要鍵必須能夠和羰基的π軌道重疊。
如果一個不對稱酮與鹼反應,存在產生兩種區域異構烯醇的傾向(忽略烯醇本身的立體化學)。例如:
三取代烯醇被認為是動力學控制的烯醇,而四取代烯醇則被認為是熱力學控制的烯醇。氫原子被去質子化后形成的動力學烯醇相對位阻更小,去質子化過程更快。總體來說,四取代烯烴由於超共軛,穩定性比三取代的烯烴更好。烯醇區域異構體的比例很大程度上受到反應中鹼的影響。以上述為例,動力學控制可以通過二異丙基氨基鋰在下得到,且具有99:1的動力學選擇性;而熱力學烯醇可以通過三苯基甲基鋰在室溫下得到10:90的熱力學選擇性。
一般來說,動力學烯醇更傾向於在低溫下形成相對離子化的金屬-氧鍵,而反應中快速去質子化需要通過使用略過量的鹼,該鹼需是強且高位阻的鹼。體積大的鹼只會中和位阻小的氫原子,低溫條件和過量的鹼能防止在初步烯醇化化時平衡轉化為更穩定的烯醇式。熱力學烯醇更傾向於長的平衡時間與更高的溫度,這種條件形成了相對共價的金屬-氧鹼而且使用少量的強鹼。不足量的鹼能夠同時去質子化所有的羰基分子,不同的烯醇負離子和羰基之間進行質子交換並形成平衡,從而形成更穩定構型。這種選擇性控制可以通過挑選金屬離子和溶劑來進行。
關於不同條件下烯醇化的擴展研究已經開始。當代有機化學在大多數情況下,已經可以得到想要的烯醇構型:
醛酮羰基旁的氫一般pKa為23左右。為了避免自身發生羥醛加成則需要使用強鹼如二異丙基氨基鋰,才可以迅速地使整個體系完全烯醇化。由於使用強鹼,且提高去質子化的選擇性,反應常在低溫下,如:丙酮乾冰浴中,於非質子極性溶劑,如在四氫呋喃、二氯甲烷等溶劑內進行。由於二異丙基氨基鋰(LDA)這類強鹼屬於硬鹼,所以這種方法叫硬式烯醇化。如果使用路易斯酸先與醛酮羰基絡合,則能極大地提高氫的酸性,用弱鹼,如:三乙胺即能實現脫質子烯醇化。相應地這種方法稱為軟式烯醇化。
烯醇或烯醇鹽相當於烯烴的衍生物而存在順反異構體。一般用E來表示反式,Z來表示順式。見圖:
不過這個命名法與烯烴體系有所區別,只要是羰基氧原子烯醇化后與雙鍵另一端的大基團處在一端的都認為是順式,而與另一個R'的基團大小無關。如:
圖例之烯醇鹽按照普通規則屬於反式,因硫的原子序數比氧大而基團優先,但在羥醛反應的研究中屬於順式。
對於酮,大多數的烯醇化條件都是得到Z構型的烯醇。而對於酯,大多數的烯醇化條件得到的是E式的烯醇。在加入六甲基磷醯胺后,該去質子化過程的立體選擇性可以與上述相反。
烯醇形成之立體選擇性可用Ireland模型進行解釋,雖然這種模型的準確性還存在疑問,如在大多數情況下無法得知在中間體是單體或是低聚體;即使如此Ireland模型仍然是理解烯醇化過程的有用工具。
在Ireland模型中,假定去質子化過程是通過六元環過渡態。兩個親電取代基團當中偏大的基團(在例子當中,甲基比氫原子更大)在過渡態中傾向於採取平伏鍵的位置,導致優先得到E構型的烯醇。在以下情況模型失效:溶劑體系從四氫呋喃變為的六甲基磷醯胺-四氫呋喃會造成烯醇的立體化學發生逆轉。
羥醛反應非常重要,因為其過程能產生兩個手性中心。許多相關研究已經了解了反應機理並能通過不同的條件改進反應的選擇性。順式/反式的轉化通常使用和碳原子上的相對構型來表示。
以前的文獻曾經使用過赤式/蘇式來命名一些碳水化合物的立體構型。
有機化學家們一直感興趣的問題,就是如何控制羥醛反應,使四種可能產物中的一種成為主要產物,並盡量減少其他異構體的生成。
每一個烯醇的雙鍵構型都確定了主要產物的相對立體構型,E型雙鍵得到反式產物;Z型雙鍵得到順式產物:
因為烯醇鹽的順反異構對羥醛產物的立體化學有著直接的影響,所以需要對烯醇鹽的順反異構進行控制,以期獲得特定結構的烯醇鹽。在胺類鹼的作用下,烯醇化的結果傾向於生成反式烯醇鹽,如果在反應體系內添加六甲基磷醯胺(),則可以使上述結果相反。
生成反式烯醇鹽的六元環過渡態大取代基間相互積壓程度小、能量低,容易形成過渡態,進而得到反式烯醇鹽;同理,生成順式的過渡態存在大取代基1,3-豎鍵相互左右,擠壓程度高、能量上不利於形成過渡態,故順式含量低。
羥醛反應研究的發展產生了更為可靠的烯醇化技術,通過選擇不同的鹼和路易斯酸催化劑,可以有效地產生以上的單一烯醇鹽。基本上而言,使用小位阻硼硬路易斯酸三氟甲磺酸二正丁基化硼()和大位阻鹼N,N-二異丙基乙基胺()有利於生成順式烯醇鹽;反之,大位阻軟路易斯酸氯代二環己基硼烷()和小位阻鹼的搭配則有利於生成反式烯醇鹽。
烯醇金屬離子的在確定羥醛反應的立體選擇性上具有很重要的作用。硼試劑常被使用,因為其鍵長遠比其他的金屬離子(如鋰,鋁或者鎂)要短。
硼-碳和硼-氧鍵的鍵長分別為1.4–1.5和1.5–1.6。而典型的金屬-碳和金屬-氧鍵的鍵長分別為1.9–2.2和2.0–2.2。硼試劑讓金屬原子“收緊”過渡態而讓反應具有更高的立體選擇性。這樣,上述反應使用烯醇鋰負離子得到的“順:反”比例為80:20;而使用二丁基硼烯醇則得到了97:3的高選擇性。
羥醛反應可以發生“底物-控制的立體化學控制”,也就是存在手性的底物可以影響到反應產物的手性。若烯醇底物含有一個位的手性中心,則可以達到完美的立體化學控制。
E式的烯醇,其主要控制因素為1,3-烯丙位張力;而對於Z式烯醇,主要控制因素這是防止1,3-位的雙直立鍵相互作用。
為了更加清晰,烯醇的立體化學被差向異構化;而在現實中同樣可以得到醛的差向異構體。在兩個例子中,1,3-順式的非對映體都是有利的。
當烯醇進攻的底物醛具有一個位的手性中心,同樣可以達到完美的立體化學控制。大多數的研究表明E式烯醇參與了Felkin非對映體的選擇;Z式烯醇參與了反-Felkin的選擇性。
由於Z式烯醇必須通過一種含有不穩定的順式-戊烷中間體或者反-Felkin旋轉異構體進行反應,所以Z式烯醇在這個例子當中降低了非對映選擇性。
若烯醇和醛都具有預先存在的手性,那就可以使用一個統一的立體化學模型來預測一個“雙重手性區分”的羥醛反應,該模型需要同時考慮:烯醇的空間影響,烯醇的立體化學,以及醛的空間影響。
現代有機合成化學需要合成光學純的化合物。然而羥醛反應創造出兩個手性中心,即形成四種手性異構體:
現今已經發展出許多用於控制相對手性(如:順式或反式)和絕對手性化學(如:RS構型)的方法。
一個廣泛使用的方法為埃文斯醯基惡唑烷酮法。其由大衛·埃文斯和同事於二十世紀七八十年代發現,這種方法通過添加一個手性助劑來暫時建立手性烯醇。這種手性助劑通過非對映選擇性反應將“手性”轉移至產物。而後將助劑脫除,得到需要的手性異構體。
在埃文斯的方法中,引入的手性助劑為惡唑烷酮,而形成的羰基化合物是一種醯亞胺。許多惡唑烷酮試劑現在都已商品化,且可買到兩種對映體,其售價約為每克10至20美元。
惡唑烷酮的醯化反應操作過程簡單。Z型烯醇可以通過硼-介導的軟性烯醇化得到順式-羥醛加合物。
通常單個非對映體可以通過羥醛加成物的結晶操作獲得。儘管成本高且只能得到“順式”產物,埃文斯羥醛由於可靠性高被廣泛使用。一些斷裂助劑的方法如下:
通過構建醯亞胺結構,順式和反式選擇性的羥醛加成反應都可進行,其允許形成四種當中的三種手性組合:順式選擇性和反式選擇性:
在順式-選擇性反應中,兩種烯醇化方法都如預期的得到Z型烯醇,然而反應的手性化學並非受到惡唑烷酮而是甲基手性中心的控制。該法還允許手性選擇的組建聚酮(一類具有反合成子的天然產物)。
現代化學方法學使羥醛反應的應用更為廣泛,其常伴隨使用催化量的手性配體。當反應使用少量的光學純配體,則可誘導光學純產物的形成,該反應稱作“催化的非對稱”反應。現下列催化非對稱反應就可實現:
前文所述手性助劑的主要局限性在於:N-乙醯醯亞胺反應不具選擇性。早期的解決方法是暫時引入硫醚基團:
向山羥醛反應是在路易斯酸,如三氟化硼或四氯化鈦的催化下,硅烯醇醚對醛的親核加成反應。向山羥醛反應並不符合齊默曼-特拉克斯勒模型。卡雷拉(Carreira)發現了一種很有用的利用硅烯酮縮醛的不對稱合成法,值得注意的是該反應具有高度的對映選擇性及廣泛的底物適用性。
該方法常應用於非支鏈脂肪族醛,由於在對映面兩側的電性和手性差別較小,該底物的親電性對於催化不對稱反應的通常較弱。
乙烯類似物(Vinylogous)的向山羥醛過程還可以形成催化非對稱性反應。下圖展示了僅對芳香族醛的有效反應,其機理被認為與具手性且金屬-鍵合的二烯醇有關。
近期發現一種新型埃文斯助劑:克里明斯(Crimmins)噻唑硫酮,能夠使反應的收率、非對映選擇性和對映選擇性都大大提高(雖然不及埃文斯的例子)。不同於早期的埃文斯助劑,噻唑硫酮對乙醯羥醛反應同樣有效,且可以通過使用少量(-)-金雀花鹼得到“埃文斯順式”或“非-埃文斯順式”加成物。該反應被認為是通過形成鈦-鍵合六元環過渡態進行的,而該過渡態類似於埃文斯助劑。
羥醛反應的另一個最新進展使用了手性二級胺催化劑進行反應。這種二級胺和酮反應會短暫形成烯胺,並與合適的醛進行對映選擇性加成。胺與羰基化合物反應得到的烯胺(類似於烯醇)可作為親核試劑參與反應,而後胺從產物上脫除重新進入催化循環。烯胺催化法總體都基於一些有機小分子,因此是一種有機催化。在下例中,脯氨酸對於催化三酮的環化反應非常有效:
該反應即Hajos-Parrish反應(同樣還稱為Hajos-Parrish-Eder-Sauer-Wiechert反應)Hajos-Parrish條件僅需要催化量的脯氨酸()。該反應沒有非手性反應的問題,因為過渡態的烯胺中間體比酮(前體)的烯醇式更具親核性。這種策略非常實用,因為它提供了一種簡單的進行對映選擇性反應的方法,而且不需要使用昂貴或有毒性的過渡金屬催化劑。
值得一提的是脯氨酸-催化的羥醛反應不具有任何非-線性效應。(線性效應是指:產物對映選擇性的比例直接與催化劑的光學純度相關)。結合同位素標記得到的證據及計算化學研究,假定的脯氨酸-催化的羥醛反應機理如下所示:
這種策略允許競爭性的雙醛交叉羥醛反應。通常兩醛之間的交叉-羥醛反應都是具有競爭性的,因為它們很容易聚合或進行非選擇性反應而得到混合產物。第一個例子如下:
基於烯醇的羥醛加成反應會優先形成順式產物,與此相反的是,有機催化形成的加成產物是反-選擇性的。在許多實例中,有機催化條件可以非常溫和而防止多聚化。然而由於兩種親電試劑都能產生烯醇化質子,為了提高選擇性就必須使用注射泵緩慢滴加親電試劑進行控制。如果其中的一種原料不具烯醇化的α氫原子或β側鏈,就可以實現可控制的加成反應。
麥克米倫和同事於2004年發現了一個很好的展示不對稱有機催化羥醛反應的例子,他們通過不同的方法保護碳水化合物(糖類)。傳統合成己糖的方法是使用多重保護-脫保護策略,共需要8至14步完成,而有機催化法能夠作用於許多相同底物,且僅用兩步就可順利完成。該反應使用了脯氨酸-催化的α羥基醛的二聚反應,然後進行幾步向山羥醛環化反應完成合成。
α羥醛的二聚化反應要防止羥醛加成物進一步發生加成反應。早期的研究表明能夠耐受α-烷氧,或者α-硅氧的保護基適用於此反應,因為能耐受吸電子基團(如乙醯基)的醛都不具反應性。保護后的赤蘚糖產物通過向山羥醛加成,而後進行半內縮醛化化轉化為四種不同的糖分子。這需要在向山加成中進行適當的非對映體控制,並且產物硅氧碳正離子會優先環化,而不是進一步進行羥醛反應。例如,葡萄糖、甘露糖、阿洛糖都可通過此法製備:
普通的羥醛反應中,羰基化合物都是進行去質子化形成烯醇。烯醇加入到醛或者酮化合物中形成醇鹽,然後進行酸化和后處理。有一種更好的方法,理論上能夠不需要多步操作,而可“直接”進行一步反應。其想法是利用金屬催化劑在羥醛加成的過程中釋放。而問題在於反應產生的醇鹽比起始原料具有更強的鹼性。產物和金屬離子緊密鍵合,從而不讓它與羰基原料繼續進行反應。
由埃文斯發現的一種方法,是使用硅基化的羥醛加成物。一種硅試劑如三甲基氯硅烷加入反應用於代替金屬離子結合醇鹽,並允許金屬催化劑的循環利用。該法減少了反應步數,降低了試劑消耗,使反應更經濟且適用於工業領域。
一種最近由Shair發現的仿生合成法,使用β-硫酮酸作為親核試劑。其中一半的酮酸在反應中進行了脫羧。其過程非常類似於通過聚酮合成酶來應用丙二-輔酶A。示例中的手性配體為雙惡唑啉。有趣的是,芳香族和支鏈脂肪族醛在此例中無法很好的進行反應。