家族性ALZHEIMER病
家族性ALZHEIMER病
家族性ALZHEIMER病是一種進行性發展的致死性神經退行性疾病,臨床表現為認知和記憶功能不斷惡化,日常生活能力進行性減退,並有各種神經精神癥狀和行為障礙。
患病率研究顯示,美國在2000年的家族性ALZHEIMER病例數為450萬例1。年齡每增加5歲,家族性ALZHEIMER病病人的百分數將上升2倍,也就是說,60歲人群的患病率為1%,而85歲人群的患病率為30%2。如果治療方法沒有取得進展,則美國在2050年時,有癥狀的病例數預期將上升至1320萬例1。家族性ALZHEIMER病病人的治療費用非常驚人;每年支出共計839億美元(按1996年的美元計算)3。
家族性ALZHEIMER病的治療包括5個主要部分:神經保護療法、膽鹼酯酶抑製劑、採用非藥物干預和精神藥理學藥物減少行為障礙、健康維護活動、臨床醫師與家庭成員及照看病人的其他人員聯合。治療需要準確診斷,並且越來越依靠對疾病病理生理的了解。
癥狀體征
AD通常起病隱匿,為特點性、進行性病程,無緩解,由發病至死亡平行病程約8-10年,但也有些患者病程可持續15年或以上。AD的臨床癥狀分為兩方面,即認知功能減退癥狀和非認知性精神癥狀。認知功能障礙可參考痴獃部分。常伴有高級皮層功能受損,如失語、失認或失用和非認知性精神癥狀。認知功能障礙可參與痴獃部分。根據疾病的發展和認知功能缺損的嚴重程度,可分為輕度、中度和重度。
1、輕度
近記憶障礙常為首發及最明顯癥狀,如經常失落物品,忘記重要的約會及許諾的事,記不住新來同事的姓名;學習新事物困難,看書讀報后不能回憶其中的內容。常有時間定向障礙,患者記不清具體的年月日。計算能力減退很難完成簡單的計算,如100減7、再減7的連續運算。思維遲緩,思考問題困難,特別是對新的事物表現出茫然難解。早期患者對自己記憶問題有一定的自知力,并力求彌補和掩飾,例如經常作記錄,避免因記憶缺陷對工作和生活帶來不良影響,例如妥善的管理錢財和為家人準備膳食。尚能完成已熟悉的日常事務或常務。患者的個人生活基本能自理。
人格改變往往出現在疾病的早期,病人變得缺乏主動性,活動減少,孤獨,自私,對周圍環境興趣減少,對周圍人較為冷淡,甚至對親人漠不關心,情緒不穩,易激惹。對新的環境難以適應。
2、中度
到此階段,患者不能獨立生活。表現為日益嚴重的記憶障礙,用過的物品隨手即忘,日常用品丟三落四,甚至貴重物品。剛發生的事情也遺忘。忘記自己的家庭住址及親友的姓名,但尚能記住自己的名字。有時因記憶減退而出現錯構和虛構。遠記憶力也受損,不能回憶自己的工作經歷,甚至不知道自己的出生年月。除有時間定向障礙外,地點定向也出現障礙,容易迷路走失。甚至不能分辨地點,如學校或醫院。言語功能障礙明顯,講話無序,內容空洞,不能列出同類物品的名稱;繼之,出現命名不能,在命名檢測中對少見物品的命名能力喪失,隨後對常見物品的命名亦困難。失認以面容認識不能最常見,不認識自己的親人和朋友,甚至不認識鏡子中自己的影像。失用表現為不能正確地以手勢表達,無法作出連續的動作,如刷牙動作。患者已不能工作,難以完成家務勞動,甚至洗漱、穿衣等基礎的生活料理也需家人督促或幫助。患者的精神和行為也比較突出,情緒波動不穩;或因找不到自己放置的物品,而懷疑被他人偷竊,或因強烈的妒忌心而懷疑配偶不貞可伴有片斷的幻覺;睡眠障礙,部分患者白天思睡、夜間不寧。行為紊亂,常撿拾破爛、藏污納垢;亂拿他人之物;亦可表現為本能活動亢進,當眾落體,有時出現攻擊行為。
3、重度
記憶力、思維及其他認知功能皆因此受損。忘記自己的姓名和年齡,不認識親人。語言表達能力進一步退化之患者只有自發言語,內容單調或反覆發出不可理解的聲音,最終喪失語言功能。患者活動逐漸減少,並逐漸喪失行走能力,甚至不能站立,最終只能終日卧床,大、小便失禁,晚期患者可原始反射等。
是一組病因未明的原發性退行性腦變性疾病。多起病於老年期,潛隱起病,病程緩慢且不可逆,臨床上以智能損害為主。病理改變主要為皮質瀰漫性萎縮,溝回增寬,腦室擴大,神經元大量減少,並可見老年斑,神經原纖維結)等病變,膽鹼乙醯化酶及乙醯膽鹼含量顯著減少。起病在65歲以前者舊稱老年前期痴獃,或早老性痴獃,多有同病家族史,病情發展較快,顳葉及頂葉病變較顯著,常有失語和失用。
病理生理
1、AD的神經病理
腦重減輕,可有腦萎縮、腦溝回增寬和腦室擴充。SP和NET大量出現於大腦皮層中,是診斷AD的兩個主要依據。
(2)大腦皮質、海馬及皮質下神經元存在大量NFT。NFT由雙股螺旋絲構成的。主要成分是高度磷酸化的tau蛋白。
2、神經化學
3、AD的分子遺傳學
已發現AD發病與遺傳因素有關。有痴獃家族史者,其患病率為普通人群的3倍。近年發現,三種早髮型家族性常染色體顯著性遺傳(FAD)的AD致病基因,分別位於21號染色體、14號染色體和1號染色體,包括21號染色體上的APP基因,14號染色體上的早老素1恩基因及1號染色體上的早老素2基因。但需注意,此類FAD的痴獃病人,只佔所有AD患者的2%左右。此外,載脂蛋白E(apoE)基因是老年型AD的重要危險基因。APOE基因位於19號染色體,在人群中有3種常見亞型,即ε2、ε3和ε4、ε5最普遍,ε4次之,而ε2則最少。有apoEε4等位基因者,患AD的風險增加,並可時發病年齡提前。但並非所謂帶apoEε4等位基因的人都會患上AD,亦有許多AD患者是沒有ε4等位基因的,國內亦有多個報道證實apoEε4是晚髮型AD的危險因素之一。
診斷檢查
AD患者的腦電圖變化無特異性。CT、MRI檢查顯示皮質性腦萎縮和腦室擴大,伴腦溝裂增寬。由於很多正常老人及其他疾病同樣可出現腦萎縮,且部分AD患者並沒有明顯的腦萎縮。所以不可只憑腦萎縮診斷AD。Spect和正電子發射斷層成像可顯示AD的頂-顳葉聯絡皮質有明顯的代謝紊亂,額葉亦可能有此現象。AD病因未明,目前診斷首先主要根據臨床表現作出痴獃的診斷,然後對病史、病程的特點、體制檢查脊神經系統檢查、心理測查與輔助檢查的資料進行綜合分析,排出其他原因引起的痴獃,才能診斷為AD。在我國心理測查包括一些國際性的測試工具。最常用的有建議只能狀態檢查,是一個非常簡單的測試工具。此外,家族性ALZHEIMER病評定量表亦是國際通用的測試工具。在鑒別診斷方面,應注意與血管性、維生素B缺乏、惡性貧血、神經梅毒、正常壓力腦積水、腦腫瘤以及其他腦原發性病變如匹克病和帕金森病所引起的痴獃相鑒別。此外,亦要注意與抑鬱症導致的假性痴獃及譫妄相鑒別。
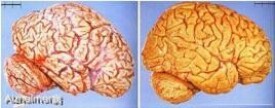
肉眼觀察腦萎縮明顯,腦回窄,腦溝寬,病變以額葉、頂葉及顳葉最明顯,側腦室及第三腦室擴張,繼發性腦積水。小腦及脊髓結構正常。
鏡下本病主要組織學病變為:(1)老年斑為細胞外結構,直徑為20~150μm,多見於內嗅區皮質,海馬CA-1區,其次為額葉和顳葉皮質。其中心為澱粉樣蛋白,周圍由銀染色陽性的神經細胞突起圍繞。其數目與痴獃成正比;(2)神經原纖維纏繞神經原纖維增粗,扭曲形成纏結,HE染色中較模糊,呈淡藍色,而銀染色最為清楚。電鏡下證實為雙螺旋纏繞的細絲構成,在海馬,杏仁核,顳葉內側的皮質錐體細胞多見。此變化是神經原趨向死亡的標誌;(3)顆粒空泡變性神經細胞漿中出現小空泡,內含嗜酸顆粒,多見於海馬區的錐形細胞;(4)Hirano小體為神經細胞樹突近端棒形嗜酸性包含體,生化分析證實為肌動蛋白,多見於海馬錐體細胞。
治療包括藥物治療與非藥物治療。認知功能障礙的藥物治療較多,但臨床療效均不確切。AD患者大腦的膽鹼乙醯基轉移酶和乙醯膽鹼酯酶活性比常人降低。有證據顯示這類神經生化改變與AD無患者的記憶損害有關係,所以AchE抑製劑可改善患者的記憶障礙。此類藥物如多那培佐,副作用較少,並無明顯肝功能異常。約1/3的AD患者治療有效,可時認知功能,但不能痊癒。膽鹼酯抑製劑石杉鹼-甲也能改善患者的記憶,副作用較少。
由於AD目前確切病因尚未得到充分闡明,治療方法主要是通過藥物作用於不同的神經遞質系統,增強中樞神經系統的高級活動,減輕疾病過程中出現的各種癥狀,延緩痴獃的進一步發展。臨床上常用的治療策略有:(1)增加腦內乙醯膽鹼(Ach)濃度的藥物;(2)促進腦內膽鹼能神經元的存活或提高其神經傳導功能的藥物;(3)減少Αβ的產生或促進其降解的藥物。
膽鹼能藥物
基於“膽鹼腦功能低下”假說,目前認為,老年性痴獃患者大腦內神經遞質Ach的缺失,是導致AD的關鍵原因,乙醯膽鹼酯酶(AchE)過多,會催化Ach的裂解反應,導致Ach缺失,神經信號傳遞失效。臨床治療AD的藥物,主要通過抑制AchE來提高患者體內Ach含量,改善AD的臨床癥狀。一般採用的方法有:(1)增加Ach合成和釋放;(2)抑制Ach降解;(3)Ach受體激動劑。目前臨床使用的治療藥物多為乙醯膽鹼酯酶抑製劑(如他克林、多奈哌齊、利斯的明、加蘭他敏、石杉鹼甲、美金剛)
抗氧化劑
司來吉蘭、維生素E、褪黑素、銀杏提取物
免疫治療
分為主動免疫治療和被動免疫治療,通過延緩和清除腦組織中Αβ的集聚,改善AD的臨床癥狀。
雌激素替代治療
腦細胞代謝激活劑
其他可能途徑
防止tau蛋白過度磷酸化
神經纖維纏結的含量與AD患者的痴獃嚴重程度密切相關,而tau蛋白異常磷酸化是神經纖維纏結產生的主要原因,抑制tau蛋白過度磷酸化可防止AD的進一步惡化。tau蛋白過度磷酸化與蛋白磷酸酶(PP)2A減少有關,可以通過提高其活性而實現治療作用。細胞周期蛋白依賴激酶5(cyclindependentkinase5,CDK5)也是一種tau蛋白磷酸化激酶,其抑製劑鈣激活中性蛋白酶有可能作為治療AD的候選藥物。
本病多發生在50~60歲,女性可能多於男性。癥狀緩慢發展,最早可為記憶力減退或其他神經衰弱癥狀,繼而出現妄想或幻覺,判斷力減退,定向力缺乏,最後痴獃,大小便失禁。家族性ALZHEIMER病
基本信息
- 傳染性
- 無傳染性