激肽釋放酶
激肽釋放酶
即徠激肽釋放酶(KLK),1930年由Kraut等在胰腺發現高濃度此物質,命名為“Kallikrein”。近30年來,隨著分子生物學和細胞生物學技術的發展和應用,發現激肽釋放酶-激肽系統(kallikrein kinin system,KKS)作為一個複雜的內源性多酶系統,參與調控心血管、腎臟、神經系統等的生理功能,與心臟病、腎病、炎症反應、癌症等疾病的發生有著密切關係。
1909年Abelous等[1]首次報道靜脈注射人尿液可引起狗的血壓短暫下降,發現尿中存在降壓物質。1930年Kraut等[2]在胰腺發現高濃度此物質,命名為“Kallikrein”,即激肽釋放酶(KLK)。在心血管系統方面的研究進展很快,許多臨床研究和基礎實驗已證實糖尿病、高血壓、心力衰竭、心肌梗死及左心室肥厚等疾病的發生與KKS的活性降低有關。因而深入研究KKS的作用為研究心血管相關疾病的發病機制和治療手段提供了又一新的途徑。
KKS是體內主要的降壓系統之一,由激肽原、KLK、激肽酶和激肽組成。激肽家族包括緩激肽(bradykinin,BK,Arg?Pro?Gly?Phe?Ser?Pro?Phe?Arg),賴氨醯緩激肽(Lys?Arg?Pro?Pro?Gly?Phe?Ser?Pro?Phe?Arg),甲硫氨醯?賴氨醯緩激肽(Met?Lys?Arg?Pro?Pro?Gly?Phe?Arg)[3]。賴氨醯緩激肽及甲硫氨醯?賴氨醯緩激肽可被血漿及尿液中的氨基肽酶降解為緩激肽。
KLK又稱血管舒緩素,是激肽系統的主要限速酶,它是一組存在於多數組織和體液中的絲氨酸蛋白酶,是一種肽鏈內切酶。它特異性的在碳末端切割底物肽,可裂解激肽原釋放具有活性的激肽,由激肽發揮對心血管系統及腎臟功能的調節作用。KLK分為兩大類:血漿KLK和組織KLK,分別由前激肽釋放酶(pre?kallikrein)和KLK前體(prokallikrein)轉換而來。它們在分子量、底物、免疫學特性、基因結構和釋放的激肽種類方面有很大差異。血漿KLK又稱Fletcher因子,特異地在肝細胞表達,是一種高分子量糖蛋白,以HMWK為底物釋放九肽即BK。組織KLK是一個大的基因家族,主要分佈在肺、腎、血管、腦、腎上腺組織,為一種中等大小的糖蛋白。在所有已知的組織激肽釋放酶家族中,只有胰/腎KLK可從激肽原釋放活性激肽,它是由KLK1基因編碼的人類KLK(hKLK1)蛋白。它主要以LMWK為底物,釋放十肽的賴氨醯緩激肽,通常稱為激肽。它的體內活性較BK強,它可以被氨基肽酶裂解為BK繼續發揮作用[4]。
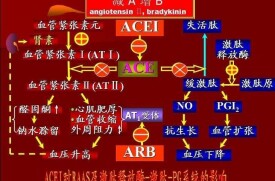
在血液和組織間隙釋出的激肽生物半衰期很短,僅有數秒,很快被激肽酶水解而失活。激肽酶主要包括激肽酶Ⅰ、激肽酶Ⅱ[即血管緊張素轉換酶(ACE)]、中性內肽酶24,11(NED24,11)、羧基肽酶及氨基肽酶。
血漿型KLK參與凝血和纖溶過程,作用於HMWK釋放BK調節血管緊張性、炎症反應以及內源性血液凝固和纖維蛋白溶解過程[5]。組織KLK分解LMWK生成激肽,參與多種生理過程,對血壓調節、電解質平衡、炎症反應等生理或病理過程進行調控[6]。
激肽主要通過自分泌和旁分泌途徑以局部激素形式與2個不同類型的BK受體即B1受體和B2受體對鄰近細胞發揮重要的生物學作用。B1和B2受體都是G蛋白耦合受體。B1受體對羧基端缺如的激肽具有高度親和力和敏感性,例如去9位精氨酸緩激肽和賴氨酸?去精氨酸緩激肽。通常認為B1受體在正常組織內缺如,主要在細菌脂多糖(內毒素)及白介素刺激和炎症時表達,可能與炎症反應和組織損傷有關。B1受體激活可刺激平滑肌細胞增殖和膠原形成,除了介導炎症介質外,還參與新生血管的形成過程[7]。B1受體在接受興奮劑后不易出現內體化和耐受,在同樣的受體密度下B1受體更依賴於基礎信號。而B2受體則存在於正常機體,密度較高,對BK和賴氨酸緩激肽敏感,一般認為B2受體介導激肽的大多數心血管效應、電解質代謝及器官保護功能[8]。BK與B2受體結合,刺激第二信使如一氧化氮(NO)/環磷酸腺苷(cAMP)和前列環素I2(PGI2)/環磷酸鳥苷(cGMP)的釋放,與腎素-血管緊張素系統(RAS)的作用相拮抗,從而發揮廣泛的生物學效應,擴張小動脈,增加局部血流,抑制腎素分泌及增加擴血管性前列腺素合酶水平,增加血管通透性及使血管舒張,促使血壓下降,調節血壓及心血管功能。
高血壓可由收縮血管物質過多或舒張血管物質缺乏引起。BK能誘導血管內皮產生舒張因子,如一氧化氮(NO)和PGI2等,從而引起擴張血管,降低外周血管阻力及調節腎臟組織對鈉鹽的排泄,參與機體血壓的調節。BK具有強大的利尿鈉效應,可使腎臟血流量增多,腎小管周圍毛細血管壓增高,抑制腎小管再吸收,並通過刺激進球小動脈壓力感受器及緻密斑而產生利尿鈉作用。另一方面,BK可抑制遠端腎小管對鈉和水重吸收及抑制抗利尿激素的作用,從而促進水鈉排泄。在原發性高血壓和腎性高血壓患者中,血管對BK的降壓反應明顯增強,從而提示高血壓狀態下缺乏內源性激肽。KKS中很多成分不足可導致BK的產生減少,從而引起高血壓。1934年德國科學家Elliot等首次發現動脈壓升高患者尿KLK排泄減少。30多年後Margolius和Sharma等肯定了這一發現並觀察到部分原發性高血壓患者或自發性高血壓大鼠(SHR)尿中KLK的水平都顯著降低,而流行病學研究也發現,尿KLK水平與原發性高血壓患者的血壓呈反比。此外,一項針對心血管危險因素的遺傳因子的研究提示腎或尿KLK基因顯性表達可降低高血壓發生的危險[9]。黑人較白人尿中KLK水平低,黑人高血壓的發病率也較白人高。對非洲裔美國黑人的研究發現伴隨著腎臟鉀排泄的減少,尿KLK排泄在高血壓前期就出現顯著降低[9]。然而鉀及醛固酮分泌水平恢復到與白種人(高加索人)相似的水平並不能使黑人的尿KLK排泄正常。這些發現提示非洲裔美國黑人KLK生物合成水平較低是由於KLK基因本身因素引起。許多動物模型也同樣能發現尿KLK排泄減少,包括SHR及Dah1鹽敏感性大鼠(DDS)。Sharma等[10]觀察到過度表達腎臟組織KLK的轉基因小鼠血壓偏低,而給予組織KLK阻滯劑抑肽酶后可使血壓恢復。激肽可降低低鹽飲食的SHR的血壓,而給予激肽B2受體拮抗劑后血壓明顯升高。激肽酶Ⅱ(ACE)抑製劑通常被應用於臨床及實驗性高血壓的治療,它的抗高血壓作用與抑制激肽的生物降解及阻斷血管緊張素Ⅱ在腎臟的形成,從而使體內激肽水平增高有關,因為同時應用激肽抗體,其降壓作用便明顯削弱。激肽原缺乏大鼠(brown Norway-Katholiek)因不能產生激肽而在接受血管緊張素Ⅱ、高鹽飲食或脫氧皮質酮和鈉鹽刺激后比正常大鼠更早出現高血壓[11]。從以上客觀事實,可以得出這樣的結論,除原發性醛固酮增多症這種因鹽皮質類固醇分泌過多的高血壓外,其餘的高血壓都可能與腎臟KKS功能低下有關。BK的降壓作用通常是經過B2受體介導的。Sharma等[12]研究發現B2受體拮抗劑B5630能夠阻斷BK的降壓作用,此外還能抑制血管緊張素轉換酶抑製劑(ACEI)卡托普利的降壓作用,由此可推斷ACEI的降壓作用除減少BK的分解而增加血液中濃度外,還能阻止激肽B2受體失敏,促使受體功能上調[13]。較早的研究顯示,口服豬胰腺KLK可明顯降低高血壓患者的血壓[14],其缺點為降壓作用短暫,須反覆給葯。給實驗動物靜脈注射提純的組織型KLK可引起快速而短暫的降壓效應[15],BK受體抑製劑艾替班特(HOE140)能阻斷此反應。已有多個應用組織KLK基因治療高血壓、逆轉左心室肥厚(LVH)、減輕腎功能損害等各種高血壓模型的研究展示轉基因治療降壓效果持久,且對心血管及腎臟疾病具有良好的保護作用。
激肽與內皮細胞B2受體內結合,釋放NO及PGI2,發揮擴張血管和抗增殖效應,保存心肌高能磷酸物,增加對糖原的攝取和利用以對抗血管緊張素Ⅱ的作用,從而發揮維持心血管內環境穩定的作用。有證據表明KKS功能失調在心力衰竭的發病機制中發揮重要作用。Whalley等[16]報道心力衰竭心臟中微血管局部激肽生成減少,NO濃度下降。此外,在起搏誘導的狗的充血性心力衰竭模型中可觀察到在使用艾替班特選擇性阻斷B2受體后冠狀動脈血流及心肌收縮力下降,左心室舒張末壓升高[17] 。因此可以認為心血管KKS活性降低促進心力衰竭的發展。另一方面,缺血預適應是一種心肌保護現象,是指心肌經1~4次短時間(2~10 min)缺血對隨後的長時間缺血性損傷產生耐受性,細胞的損傷明顯減輕。其機制至今尚未完全闡明,已有的研究證實,缺血可觸發內源性自我保護,釋放一系列內源性活性物質,BK便是其中之一。BK在缺血早期就由心肌組織釋放,局部及全身性給予外源性BK可明顯增加冠狀動脈及毛細血管血流,改善心肌代謝[18]。Scholkens[19]研究證實在狗的冠狀動脈內注入BK能顯著降低缺血誘導的嚴重心律失常。BK冠狀動脈灌注可提高結紮冠狀動脈的SHR及WKY大鼠的存活時間,且該作用可被艾替班特抵消,提示BK對缺血預適應的心臟保護作用是由B2受體介導,通過激活信號轉導途徑產生NO和PGI2實現的[20]。對大鼠、狗及人的多個研究顯示激肽在心肌及全身缺氧缺血狀態下持續釋放,特別是急性心肌梗死後,這一過程提示激肽在心肌梗死時發揮保護作用。藥理學和遺傳學研究顯示KLK和激肽受體在缺血后被誘導活化是機體的自發性反應,以增加患部血液灌注,促進康復。因此急性心肌梗死後心肌BK量增加被認為是減少或限制梗死面積、增加心臟電穩定性、抑制再灌注心律失常的有益反應。隨著分子生物學和基因定位的研究,越來越多的證據表明KKS在心血管病理生理學中的重要性,也為我們發展以KKS為基礎治療心血管疾病提供了可能。
研究認為KKS部分作用與RAS系統相反[5]。在生理條件下,內皮細胞及其基質提供絲氨酸蛋白酶PRCP,激活激肽釋放酶原(PK)轉變為KLK。隨後激肽釋放酶激活FX Ⅱ[21],在與BK的前體激肽原結合后,無活性的PK轉化為KLK。接下來,PRCP介導的向KLK的轉化使激肽原轉變為BK。BK 與內皮細胞B2受體結合,刺激細胞內Ca2+動員,引起NO和前列腺素的釋放。通過同一受體,緩激肽還增加內皮儲存池組織型纖溶酶原激活劑(t?PA)釋放[22],發揮抗凝血作用。AngII刺激內皮細胞中纖溶酶原激活物抑制1(PAI?1)mRNA的表達,以劑量依賴方式升高血漿PAI?1水平[23],發揮促進血栓形成的作用,PRCP可降解Ang Ⅱ從而使PAI?1水平降低。血漿KLK可促進單鏈尿激酶和纖溶酶原的活化,發揮抗血栓形成作用。激肽原及降解產物也具有抗凝血酶活性。BK降解產物緩激肽(1?5)能通過與蛋白酶活化受體1和4上的凝血酶裂解部位結合,抑制凝血酶誘導的血小板聚集[24]。FXⅡ,PK和HMWK是參與內源性凝血的蛋白質,Merlo等[25]採用對照的方法對200名心肌梗死患者血中FXⅡ,FXⅠ,PK和HK的含量進行測定,發現FXⅠ,HMWK和PK水平顯著升高,表明它們對心肌梗死的發生可能起一定的作用。
LVH被認為是高血壓患者的獨立危險因素。BK能夠對抗主動脈結紮引起高血壓大鼠LVH的發展,這種抗心肌肥厚的效應能被B2受體拮抗劑艾替班特殊性及NO合酶抑製劑L?NNA抵消,說明BK是通過降低NO釋放來發揮抑制LVH的作用,證實在SHR大鼠LVH的發病機制中心血管KKS的缺乏佔有重要作用,而心血管組織KLK及激肽原的活性降低是導致心臟BK減少的原因,因此心臟KKS組分的不足可能引起高血壓和LVH心肌功能障礙。在用ACEI雷米普利進行降壓及逆轉LVH的觀察中發現,大劑量雷米普利[1 mg/(kg·d)]共6周的治療可降低血壓,抑制LVH的發展,而給予小劑量雷米普利[10 μg/(kg·d)]6周后對血壓及血漿中ACE活性沒有影響,但阻止了主動脈結紮后引起的LVH。2種劑量雷米普利抗心肌肥厚的作用及大劑量雷米普利的降壓作用均可被B2受體拮抗劑艾替特及NO合酶抑制抵消。該研究說明KKS和RAS在阻止或延緩心室肥厚這一靶器官損害的重要作用,更支持了KKS是心血管保護因子的觀點。
大量研究表明KKS在心血管系統各種疾病的發病機制中發揮相當重要的作用,如高血壓、心力衰竭及心肌缺血、LVH及內皮功能紊亂。隨著人們對KKS的認識不斷深化,不但在心血管方面,而且在其他多種病理過程中的作用逐漸成為研究的熱點。特異性受體正成為研究的新靶點,相應拮抗劑的問世將成為新一代更具選擇性的治療心血管疾病、炎症、疼痛及免疫性疾病的新型藥物。
【參考文獻】
[1]Abelous JE,Bardier E. Les substnces hypotensives de lurine humaine normale [J].Compt Rend Soc Biol,1909,66(3):511?522.
[2]Kraut H,Frey EK,Werle E.Der Nachweis eines Kreislaufhormon in der Pankreasdruse[J].Hoppe?Seyler?s Z Physiol Chem,1930,189(1):97?106.
[3]Sharma JN.Does kinin mediate the hypotemsive action of angiotensin converting enzyme(ACE)inhibitors?[J]. Gen Pharmacol,1990,21(4):451?457.
[4]Yousef GM,Diamandis EP.The new human tissue kallikrein gene family: structure,function,and association to disease[J].Endocr Rev ,2001,22(2):184?204.
[徠5]Schmaier AH. The plasma kallikrein?kinin system counterbalances the rennin?angiotemsin system[J].Clin Invest ,2002,109(8):1007?1009.
[6]Diamandis EP, Yousef GM, Egelrud J, et al.New nomenclature for the human tissue kallikrein gene family[J].Clin Chem,2000,46(11):1855?1858.
[7]Pesquero JB,Araujo RC,Heppenstall PA,et al.Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 recrptors[J].Proc Natl Acad Sci USA,2000,97(14):8140?8145.
[8]Hagiwara M,Murkkami H,Ura N,et al. Renal protective role of bradykinin B1 receptor in stroke-prone spontaneously hypretensive rats[J].Hypertension Res,2004,27(6):399?408.
[9]Wong CM,OConnor DT,Martinez JA, et al.Diminished renal kallikrein responses to mineralocorticoid stimulation in Afican Americans:determinants of an intermediate phenotype for hypertension[J].Am J Hypertens,2003,16(4):281?289.
[10]Sharma JN, Amrah SS,Noor AR.Suppression of hypertensive responses of captopril and enalapril by kallikrein inhibitors aprotinin in spontaneously hypertensive rats[J].Pharmacology,1995,50(6):363?369.
[11]Majima M, Mizogami S,Kuibayashi Y, et al.Hypertension induced by a nonpressor dose of angiotensin II in kininogen?deficient rats[J]. Hypertension,1994,24(1):111?119.
[12]Sharma JN,Stewart JM,Mohsin SSJ, et al. Influence of a kinin antagonist on acute hypertensive responses induced by bradykinin and captopril in spontaneously hypertensive rats[J].Agents Actions,1992,38(Pt 3):258?269.
[13]Braunwald E. Shattuck lecture?cardiovascular medicine at turn of the millennium:triumphs ,concers and opportunities[J].N Eng J Med,1997,337(19):1360?1369.
[14]Overlack A, Stumpe KO, Kolloch R,et al. Antihypertensive effect of orally administered glandular kallikrein in essential hypertension. Results of double blind study[J].Hypertension,1981,(3Pt 2):118?121.
[15]Chao J,Stallone JN, Liang YM,et al.Kallistatin is a potent new vasodilator[J].Clin Invest,1997,10(1):11?17.
[16]Whalley ET,Solomon JA ,Modafferi DM.CP?0127, a novel potent bradykinin antagonist increases survival in rat and rabbit model of endotoxin shock[J].Agents Actions ,1992,38(3):413?420.
[17]Koide A ,Zeitlin IJ,Parratt JR.Kinin formation in ischaemic heart and aorta of anaesthetized rats[J]. J Physiol(Lond),1993,467(1):125P.
[18]Linz W, Wiemer G,Gohlke P.Contribution of kinin to the cardiovascular action of converting-enzyme inhibitors[J].Pharmacol Rev,1995,47(1):25?49.
[19]Scholkens BA. Kinins in the cardiovascular system[J].Immunopharmacology,1996,33(1?3):209?216.
[20]Abbas SA, Sharma JN, Yusof APM. The effect of bradykinin and its antagonist on survival time after coronary artery occlusion in hypertensive rats[J]. Immunopharmacology,1999,44(1/2):93?98.
[21]Joseph K, Tholanikunnel BG, Kaplan AP. Activation of the bradykinin forming cascade on endothelial cells: a role for heat shock protein 90[J]. Int Immunopharmacol,2002, 2(13/14): 1851?1859.
[22]Pretorius M,Rosenbaum D,Vaughan DE,et al. Angiotensincoverting enzyme inhibition increases human vascular tissue?type plasminogen activator release through ehdogenous bradykinin[J]. Circulation,2003,107(4):579?585.
[23]Brown NJ, Agirbasli MA, Williams GH, et al. Effect of activation and inhibition of the renin?angiotensin system on plasma PAI?1[J]. Hypertension, 1998,32(6):965?971.
[24]Hasan AA, Amenta S, Schmaier AH. Bradykinin and its metabolite, Arg?Pro?Pro?Gly?Phe, are selective inhibitors of alpha?thrombin?induced platelet activation[J].Circulation,1996,94(3):517?528.
[25]Merlo C, Wuillemin WA, Redondo M,et al. Elevated levels of plasma prekallikrein, high molecular weight kininoen and factor XI in coronary heart disease[J]. Atherosclerosis,2002,161(2):261?267.
組織激肽釋放酶的生物學性質和作用機制
人體內的激肽釋放酶包括血漿激肽釋放酶和組織激肽釋放酶,二者分別由前激肽釋放酶(prekalikrein)和激肽釋放酶原(prokallikrein)轉換而來。血漿激肽釋放酶催化高分子激肽原水解,生成緩激肽(bradykinin)和胰激肽(kallidin)。在人體內,組織激肽釋放酶又稱為胰/腎激肽釋放酶[4],它能催化低分子激肽原水解,生成胰激肽。緩激肽和胰激肽在激肽酶I的作用下羧基端水解掉Arg,分別生成des-Arg_-BK和des-Arg_-kallidin,後者仍具有生物活性,需要血管緊張素轉化酶或氨基肽酶才能完全滅活,激肽主要與G蛋白耦聯的B1 R,B2 R結合發揮作用。B2 R為管家基因表達,是正常狀態下激肽發揮作用的主要受體,對緩激肽,胰激肽敏感;而B1R在炎症和缺血等損傷下誘導生成,對des-Arg_-kallidin,des-Arg -BK敏感,其中B1 R對des-Arg_-kallidin的敏感度大於des-Arg -BK。認為B1R可能參與損傷部位的炎症反應和循環改善,並在新生血管生成中起重要作用。激肽與受體結合后,激活NO-CGMP和PG-CAMP途徑,從而調節NO和PG等生物活性物質的釋放來參與多器官功能調節和多種病生過程,如抑制凋亡、炎症、肥大、纖維化,促進心腎腦血管的新生血管的生成和腦的新生神經的生成。
組織激肽釋放酶在心血管及腎臟的保護作用
人組織激肽釋放酶(HTK)廣泛存在於人腎、心血管、中樞神經系統、胰、腸等臟器中,並通過其代謝產物與受體結合,來發揮其廣泛的病理生理作用。其中以HTK 在心血管及腎疾病方面的研究最多。
激肽釋放酶-激肽系統(kallikreinkinin system ,KKS)在維持正常血壓,保護心臟方面起重要作用,激肽釋放酶-激肽系統(kallikreinkinin system ,KKS)缺陷會引起高血壓,Berry在1989年對一份家族進行的研究顯示人尿激肽釋放酶(human urinary kallikrein, HUK)可減少高血壓的風險。許多高血壓或心肌缺血再灌注(I∕R)模型的動物實驗表明,以腺病毒為載體的人組織激肽釋放酶基因(ad. htk)轉導能降低高血壓,緩解心肌肥厚和纖維化,還可提高心臟功能,減少心肌梗死範圍,減少心肌I∕R后的室顫和凋亡。
人尿激肽釋放酶(human urinary kallikrein, HUK)是一種顯著的腎血管擴張劑,利尿劑,促鈉排泄劑,能保護腎臟。人尿激肽釋放酶(HUMAN URINARY KALLIKREIN, HUK)下降會引起住院病人輕度腎病,嚴重者可引起嚴重腎衰,激肽釋放酶-激肽系統(kallikreinkinin system ,KKS)可通過抑制炎症和氧化酶來對抗高鹽飲食或藥物引起的腎衰。
組織激肽釋放酶對腦組織的保護作用
在人類,已證實組織激肽釋放酶分佈在丘腦、下丘腦、腦灰質、腦幹網狀結構的神經元和腺垂體細胞及脈絡叢細胞上。B2R在人星形神經膠質、少突膠質細胞、小膠質細胞、腦血管內皮細胞、大腦皮質、紋狀體、丘腦、下丘腦的神經元上都有表達。而B1R在丘腦、下丘腦的神經元和基底動脈中有表達。體外研究顯示人類B1R在血管內皮細胞、大動脈的平滑肌細胞、冠狀動脈、肌性小動脈中都有存在。在缺血等損傷或炎症時,B1R表達上調。這些都為組織激肽釋放酶通過代謝產物激肽結合B1R和B2R來保護腦組織提供了前提,具體的神經保護作用及其作用機製表現如下:
1 擴張腦動脈,改善缺血腦組織血供和氧供
人們對腦缺血的病理生理進行了深入研究,並提出了多種學說,但迄今為止沒有一種機制能完全闡明腦缺血的損傷機制。現認為參與腦缺血損傷的分子機制有興奮性氨基酸的釋放、鈣離子穩態失衡、自由基的形成、蛋白酶的激活及NO的介導作用等。
NO在腦缺血損害中所起的作用一直是研究的熱點。NO具有神經保護和神經毒素雙重作用。認為NO的雙重作用與其產生來源有關,NO由NOS 催化底物L - 精氨酸合成。NOS 可分為結構型(cNOS) 和誘導型(iNOS) ,cNOS 包括內皮源性(eNOS) 和神經源性(nNOS) 。實驗證明,源於iNOS和nNOS過度表達所形成的NO有神經毒性,而源於eNOS所產生的NO卻有神經保護作用。所以能通過上調eNOS來增加NO的藥物能起神經保護作用。
B1R和B2R是特殊的可調節的G蛋白耦聯受體,已證實它們在內皮細胞中的細胞信號轉導通路相同。當激肽與B1 R或B2 R結合后,受體胞內端耦聯的G蛋白激活磷酸酯酶C(PLC)激活,PLC進一步水解4,5-二磷酸肌醇(IP3),IP3彌散到胞漿中與肌漿網中IP3受體結合,引起貯庫中Ca2+釋放,細胞外Ca2+內流,使細胞內Ca2+增加,最後激活eNOS,產生NO。胞內Ca2+增加還激活磷脂酶A2(PLA2),誘生PGI2。Lamontagne,Be´lichard 等指出des-Arg_-BK擴血管作用至少部分由NO介導,PG似乎不重要。激肽擴張腦動脈的作用部分來自NO的釋放。
急性中風病人在缺血發作起始后8天期間外周循環中胰激肽升高。Simone等研究顯示:22例較大梗死灶的大腦中動脈閉塞患者較14例正常成人,其組織激肽釋放酶濃度呈升高趨勢,胰激肽濃度升高。這些都說明在缺血腦組織中,組織激肽釋放酶和胰激肽激活。胰激肽具有明顯的擴張血管作用。胰激肽及其代謝物des-Arg_-kallidin能分別與B2R,B1R結合併釋放NO,擴張腦動脈。在正常人和動物,擴血管作用主要由B2R介導;而在炎症或缺血等損傷情況下,主要由新表達的B1R介導擴血管作用。如在病理情況下,B1R顯示出比B2R更明顯的擴張冠脈的作用。
在急性腦梗死等缺血性損傷時,缺血部位血管細胞被誘導生成B1R,此時激肽即與B1R結合擴張缺血區腦組織的動脈血管,從而改善缺血腦組織血供和氧供。
2促進缺血腦組織的新生血管的生成
在外周血管病的病人和動物模型中顯示激肽釋放酶-激肽系統(KALLIKREINKININ SYSTEM ,KKS)上調,激肽釋放酶-激肽系統(KALLIKREINKININ SYSTEM ,KKS)在心肌/四肢缺血性疾病中的促進新生血管形成和抑制細胞凋亡中起重要作用。有理論認為,激肽通過增強血管形成對缺血組織存在長時間的保護作用。局部轉導HTK基因能引起該區血管生成和促進組織恢復。活體實驗表明,HTK 基因轉導能促進兔角膜新生血管的生成和毛細血管增生。有研究表明,低劑量(106 PFU)的ad. htk 轉導入小鼠能促進四肢肌肉毛細血管、動脈的生長,107 PFU的ad.htk可使微血管進一步擴張。對鏈脲黴素引起的糖尿病小鼠,局部予KLK能使其後肢骨骼肌微血管減少的進程中止。這一作用是通過抑制凋亡,促進血管再生來實現的。運用組織激肽釋放酶抑製劑KLK結合蛋白抑制KLK后,可觀察到其抑制毛細血管內皮的增值並誘導其凋亡,最終抑制新生血管的形成。
在體外研究中發現,激肽通過IP3-AKt/蛋白激酶B(即IP3-AKt-B)或鈣調蛋白途徑激活內皮一氧化氮合酶(eNOS),從而使血管內皮生長因子受體通過eNOS介導引起基質層內皮細胞形成。體內研究表明,AKt-B和eNOS與ad.htk引起的新生血管形成通路是功能相關的。ad.htk誘導生成的激肽與血管內皮生長因子A共同發揮以下作用:誘發血管生成,產生NO,舒張血管。
藥理學研究表明:B1R在毛細血管增殖方面起作。B1R在缺血性損傷中不僅直接調解內皮細胞的生長和存活(激肽能有效吸引白細胞,白細胞是產生內皮細胞生長因子需要的),還能通過增加血管外血漿蛋白的滲出來參與缺血后新生血管的生成(這些蛋白為血管形成提供臨時支架)。新近有研究表明:遺傳性B1R缺乏則修復性的新生血管就不能形成。所以,B1R在促進缺血組織的新生血管的生成中起重要作用。
在急性腦缺血和細胞損傷時,缺血部位細胞B1R表達上調,組織激肽釋放酶通過代謝產物des-Arg_-kallidin與B1R結合,並進一步通過IP3-AKt-B或鈣調蛋白途徑激活內皮一氧化氮合酶(eNOS),從而促進缺血腦組織的新生血管的生成。
3促進神經膠質細胞遷移和抑制細胞凋亡,減少炎症細胞的侵潤
不同的動物模型都表明組織激肽釋放酶(KLK)通過抑制細胞凋亡和炎症細胞侵潤來減少心腎腦等器官損傷。Julie Chao和 Lee Chao在阻斷大腦中動脈(MCAO)造成大鼠局部腦缺血的模型中,將ad.htk導入腦室后,能明顯減少缺血誘導的神經功能損傷,縮小腦梗死面積,促進神經膠質細胞存活和遷移至缺血性半影區和中心,減少神經細胞和神經膠質細胞的凋亡,炎症細胞的侵潤,促進新生血管生成和神經細胞再生,從而提高了存活率。腦MCAO后靜脈持續微泵輸入人組織激肽釋放酶對缺血再灌注引起的行動障礙等神經功能恢復有直接作用。
MCAO引起的腦缺血再灌注破壞了血腦屏障,KLK基因或蛋白可通過血腦屏障進入腦缺血損傷區,進而發揮神經保護作用。形態學分析顯示:KLK基因轉導增強了存活率,促進神經膠質細胞遷移至缺血性半影區和中心。KLK基因導入后細胞存活率提高與與升高腦中NO水平和phospho-Akt,Bcl-2水平,減少半胱天冬酶-3活化,降低NAD(P)H氧化酶活性,抑制超氧化物產生有關。這說明KLK基因或蛋白轉入對腦缺血性損傷的保護作用不依賴於其擴血管,降血壓作用,而是通過以下途徑:促進神經膠質細胞存活和遷移,通過抑制氧化應激和抑制Akt-Bcl-2信號轉導通路來抑制凋亡。
研究顯示激肽在細胞遷移和凋亡方面的效應能被B2 R拮抗劑艾替班特阻滯,表明B2 R介導這些作用,B2 R在保護缺血腦卒中的作用同時也在B2 R缺陷鼠中得以證實。B2 R缺陷鼠在缺血再灌注(I/R)損傷后比野生型鼠表現出更明顯的梗死範圍和凋亡,更嚴重的行動障礙。它們在缺血第1天白細胞聚集比野生型鼠減少,但在缺血第3天白細胞聚集比野生型鼠多。有研究顯示早期(腦缺血0.25 h~6.25 h)用B2 R拮抗劑可通過抑制水腫形成來減弱一過性腦缺血損傷。以上表明B2R有雙重的效應:缺血早期促進炎症細胞的侵潤,引起血管滲透性增加,但晚期通過促進Akt磷酸化,降低NAD(P)H氧化酶活性,抑制超氧化物起神經保護作用。
4.拮抗血管損傷,抑制動脈肥厚
Murakami等在給行血管球囊成形術后鼠左頸總動脈局部轉導KLK基因后,動脈內膜/中膜比值明顯比對照組低,這一作用被NOS抑製劑L-NAME拮抗,說明它是NO依賴性的。Emanueli等在鼠動脈重構模型中,發現全身給KLK基因通過改變血管剪切應力來減少新內膜形成。zhang等在高鹽飲食引起的鼠高血壓腦出血模型中轉導KLK基因后,顯著緩解高血壓,抑制動脈肥厚,減輕腦血腫,正是通過以上作用,組織激肽釋放酶在高血壓腦出血中起到了重要保護作用,從而使動物死亡率下降。
基本信息
- 中文名
- 激肽釋放酶