共找到2條詞條名為右佐匹克隆的結果 展開
- 一種化合物
- 艾司佐匹克隆
右佐匹克隆
一種化合物
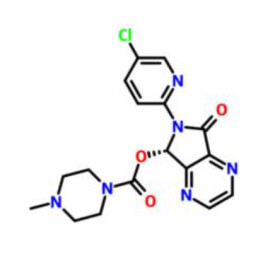
右佐匹克隆(eszopiclone),是一種有機化合物,化學式為C17H17ClN6O3,呈白色或略淡黃色粉末。
右佐匹克隆(eszopiclone)白色或略淡黃色粉末,分子式為C17H17ClN6O3,分子量為388.80800,密度為1.54 g/cm3,熔點為202-204ºC,沸點為580.7ºC at 760 mmHg。
本品應個體化給葯,成年人推薦起始劑量為入睡前2mg,由於3mg可以更有效的延長睡眠時間,可根據臨床需要起始劑量為或增加到3mg。
主訴入睡困難的老年患者推薦起始劑量為睡前1mg,必要時可增加到2mg,睡眠維持障礙的老年患者推薦劑量為入睡前2mg(見注意事項)。
如高脂肪飲食后立刻服用右佐匹克隆有可能會引起藥物吸收緩慢,導致右佐匹克隆對睡眠潛伏期的作用降低(見葯代動力學)。
特殊人群:嚴重肝臟損患者應慎重使用本品,初始劑量為1mg。
合用CYP抑製劑:與CYP3A4強抑製劑合用,本品初始劑量不應大於1mg,必要時可增加至2mg。
根據國外臨床試驗的報道:
右佐匹克隆上市前研究中,大約有400名正常受試者參加了臨床藥理/葯代動力學研究,大約1500名患者參加了安慰劑對照的臨床有效性研究(相當於大約263個患者暴露年)。上市前研究中右佐匹克隆治療的條件及療程差異較大.包括開放試驗和雙盲試驗,住院病人和門診病人,長期和短期試驗,不良反應是通過收集不良事件,評估體格檢查、生命體征、體重、實驗室檢測,ECG等結果來進行評價的。
通過問診和臨床研究者使用自己選擇的專業術語記錄不良事件。下面的表格中使用COSTART術語分類報道不良反應。
安慰劑對照的試驗中觀察到的不良反應
導致停葯的不良事件
對老年人進行的安慰劑平行對照試驗中,208名使用安慰劑患者中有3.8%,215名服用2mg右佐匹克隆患者中有2.3%,72名服用1mg右佐匹克隆的患者中有1.4%均由於不良事件停止治療。在對成年人的6周平行組對照研究中,3mg右佐匹克隆沒有一例因為不良事件停止治療。在對成人失眠患者的6個月長期研究中,195例服用安慰劑患者中有7.2%,593例服用3mg右佐匹克隆患者中有12.8%由於不良事件停止試驗。而非發生不良事件原因退出試驗的發生率高於2%。
對照試驗中發生率大於2%的不良事件
表1顯示一項為期44天的Ⅲ期安慰劑對照的臨床試驗中,成年患者服用2mg或3mg右佐匹克隆,與治療相關的不良事件的發生率。本表中僅包括與安慰劑組相比,2mg或3mg右佐匹克隆組大於2%的不良事件,
表1:成年患者服用右佐匹克隆共6周的安慰劑對照的臨床試驗中與治療相關的不良事件的發生率
註:與安慰劑組相當或發生率小於安慰劑組的不良事件包括異常夢境、意外創傷,胃痛、腹瀉、流感、肌痛、疼痛、喉痛、鼻炎,
表1可見成年患者與劑量相關的不良事件包括病毒感染、口乾、眩暈、幻覺、感染、皮疹,味覺異常,其中味覺異常的劑量相關性最明顯。
表2顯示老年患者(年齡65-86歲)服用1mg或2mg右佐匹克隆14天的聯合Ⅲ期安慰劑對照的臨床試驗中,與治療相關的不良事件的發生率,表中僅包括與安慰劑組相比,1mg或2mg右佐匹克隆組不良事件大於2%的事件。
表2老年患者(年齡65-80歲)服用右佐匹克隆2周的安慰劑對照的臨床試驗中與治療相關的不良事件的發生率
註:與安慰劑組相當或發生率小於安慰劑組的不良事件包括腹痛、衰弱、噁心、紅疹、嗜睡。
表2可見老年患者中與劑量相關的副反應包括:疼痛、口乾、味覺異常,其中味覺異常與劑量相關性最為明顯。
由於臨床試驗中患者特徵及其他因素的不同,這些數據不能用來預測正常醫療實踐中不良事件的發生率。同樣,由於治療藥物、使用方法與臨床評價者的不同,本試驗中所引用的數據與其他臨床試驗的數據是不可比的。但是這些數字可以為臨床醫生提供一些參考。
對本品及其成份過敏者,失代償的呼吸功能不全患者,重症肌無力、重症睡眠呼吸暫停綜合症患者禁用。
特別注意
由於睡眠障礙可能是生理和/或心理紊亂的表現,僅有在仔細對患者進行評價後方採取對症治療。7-10天治療后若失眠仍然出現則表明存在原發性心理和/或醫學疾病。失眠的惡化或出現新的想法及行為的異常都有可能是束被認知的心理或生理障礙的結果。鎮靜/催眠藥物,包括右佐匹克隆治療期間有可能出現上述情況。由於右佐匹克隆的一些副反應是劑量相關的,使用最低有效劑量是非常重要的,尤其對老年患者。
有報道,服用鎮靜/催眠藥物會產生一系列的異常想法和行為改變。有一些變化類似於酒精及其他中樞神經系統抑製劑的作用,例如進攻性及與性格不符的外向。其他有報道的行為變化包括行為奇怪、激動、幻覺和失去人格,不可預見的有可能出現健忘和其他神經精神的癥狀。抑鬱的患者服用鎮靜/催眠藥物有抑鬱加重、包括出現自殺想法的報道。
根難確定上述的異常行為是否是藥物引起的、自發的或心理或生理紊亂的結果。然而,任何新的行為體征或癥狀的出現均應仔細評價。
使用鎮靜/催眠藥物荊量快速下降或突然停葯時,有可能與其他CNS抑製劑出現類似的戒斷體征或癥狀。
與其他催眠藥物一樣,右佐匹克隆有中樞抑制作用。由於快速起效,右佐匹克隆應僅在上床準備睡覺前服用或已經上床但睡眠困難時服用。在服用該藥物后及第二天,患者應小心從事包括需要完全警覺或行為協調等危險性的工作(例如,操作儀器或開車)。與其他催眠藥物一樣,右佐匹克隆與其他精神科藥物、抗驚厥藥物、抗組胺藥物、乙醇和其它產生CNS抑制作用的藥物合用可能產生額外的CNS抑制作用。右佐匹克隆不可與酒精同服。由於可能產生的相加作用,右佐匹克隆與其他CNS抑製劑舍用應進行劑量的調整。
服藥時間
右佐匹克隆應在臨睡前服用。服用鎮靜/催眠藥物有可能產生短期記憶損傷、幻覺、協調障礙、眩暈和頭暈眼花。
老年和/或虛弱患者使用
老年患者和/或虛弱患者使用鎮靜/催眠藥物應考慮到重複使用或對藥物敏感引起的運動損傷和/或認知能力損傷。對於此類患者推薦起始劑量為1mg。
伴有其他疾病的患者
對伴有其他疾病的患者服用右佐匹克隆的臨床經驗有限。有可能對代謝或血液動力學造成影響的疾病服用右佐匹克隆應注意。
對健康志願者進行的一項臨床研究中,服用推薦劑量2倍(7mg)的右佐匹克隆不產生呼吸-抑制作用。但是若患有呼吸障礙疾病的患者使用右佐匹克隆,建議引起注意。
患有嚴重肝損傷患者由於系統暴露量為正常肝功能患者的2倍,服用右佐匹克隆劑量應降低到1mg。對於輕微或中度肝功能損傷患者沒必要進行劑量調整.由於小於10%的右佐匹克隆通過尿液以原形藥物代謝,腎功能損傷患者沒必要進行劑量調整。當與CYP3A4強抑製劑,如酮康唑合用時,應降低右佐匹克隆的劑量。右佐匹克隆與有CNS抑制作用的藥物合用時也建議減小劑量。
抑鬱患者使用
顯示抑鬱癥狀的患者應小心服用鎮靜/催眠藥物。對於此類患者有可能出現自殺傾向,有可能需要保護。這類患者常見故意過量服用藥物,因此,每次處方量應儘可能的最低。
本品由於具有適當的親脂性,容易進入大腦,右佐匹克隆及其代謝產物可部分通過胎盤屏障,同時本品在乳汁中濃度可能較高,因此妊娠婦女及哺乳期婦女慎用此葯。
有關18歲以下兒童用藥的安全性、有效性尚未確立,不推薦服用此葯。
用藥時,可先從小劑量開始逐漸增量,以便得到適合於患者的劑量,參見【用法用量】項。
具CNS活性藥物
乙醇:右佐匹克隆與0.70g/kg乙醇合用可對神經運動功能產生相加作用影響,可持續4小時。
帕羅西汀:每天合用3mg右佐匹克隆及2mg帕羅西汀,共7天,無葯代動力學及藥效間的相互作用。
勞拉西泮:合用3mg右佐匹克隆及2mg勞拉西泮無臨床相關性的藥效及葯代動力學的影響。
奧氮平:合用3mg右佐匹克隆及10mg奧氮平使DSST評分降低。相互作用為藥效的改變而非葯代動力學的改變。
抑制CYP3A4的藥物(酮康唑)
CYP3A4是右佐匹克隆消除的主要代謝通道。與400mg酮康哇(一種CYP3A4的強抑製劑)合用5天可使右佐匹克隆AUC增加2.2倍。Cmax和t1/2分別增加1.4倍和1.3倍。其他CYP3A4的強抑製劑可能產生相似的作用(例如:伊曲康唑、克拉黴素、奈法唑酮、竹桃黴素、利托那韋、奈非那韋)。
誘導CYP3A4的藥物(利福平)
與CYP3A4的強誘導劑利福平合用可使消旋佐匹克隆暴露率降低80%。右佐匹克隆可能產生相似的作用。
血漿蛋白結合力強的藥物
右佐匹克隆血漿蛋白結合率不是根強(52%-59%);因此,右佐匹克隆的分佈不應對蛋白結合敏感。患者服用3mg右佐匹克隆及蛋白結合力強的藥物不應該改變兩種藥物的遊離濃度。
治療指數窄的藥物
地高辛:服用地高辛第一天0.5mg一天兩次,隨後6天每天0.25mg不影響單劑量3mg右佐匹克隆的葯代動力學參數。
華法林;服用3mg右佐匹克隆5天不影響(R)與(S)華法林的葯代動力學參數:口服25mg華法林,不影響右佐匹克隆的藥效學參數。
大劑量使用右佐匹克隆上市前臨床試驗是有限的。右佐匹克隆的臨床試驗中報道了一例使用36mg超劑量右佐匹克隆的患者完全康復。使用消旋佐匹克隆超劑量340mg的患者也完全康復(相當於右佐匹克隆最大推薦劑量的56倍)。
癥狀和體征
臨床前藥效作用的放大可被認為是服用中樞神經系統抑製劑過量的癥狀和體征。意識損傷的程度從嘻睡到昏迷不醒等。消旋佐匹克隆在歐洲上市后,曾有超劑量致死的報道,但這類事件多與其他中樞神經系統抑製劑合用。
超劑量后推薦的治療方法
儘快洗胃、對症及支持治療。必要時靜脈補液,氟馬西尼可能有用。在所有超劑量使用藥物的病例中,應對病人的呼吸、脈搏、血壓進行監涮,同時採用一些全身性支持療法,對低血壓和中樞神經系統抑制的病例應該進行監測和採取相應的治療措施。透析法的價值未知。
1.毒理研究:
遺傳毒性
右佐匹克隆小鼠淋巴瘤細胞染色體畸變試驗結果陽性、CHO細胞染色體畸變試驗結果不明確,Ames試驗、UDS試驗,小鼠搬核試驗結果均為陰性。右佐匹克隆代謝產物(S)-N-脫甲基-佐匹克隆CHO細胞、人淋巴細胞染色體畸變試驗結果為陽性,Ames試驗、32P-末端標記DNA加合試驗、小鼠在體骨髓細胞染色體畸變試驗、微核試驗結果均為陰性。
生殖毒性
在生育力與早期胚胎髮育毒性試驗中,雄性與雌性大鼠經口給予右佐匹克隆分別達45、180mg/Kg/天,兩種性別動物的生育力均降低,雌雄動物在高劑量給葯時,雌性動物未發生妊娠,未見影響劑量均為5mg/Kg/天(按mg/m推算,相當於人最大推薦劑量的16倍)。其他影響包括著床前丟失增加(無影響劑量為25mg/Kg)、動情周期異常(無影響劑量為25mg/Kg),以及精子數量與活動度降低、形態異常精於數增加(無影響劑量為5mg/Kg)。
在胚胎-胎仔發育毒性試驗中,妊娠大鼠與家兔在器官形成期經口給葯,在所測試的最高劑量下未見致畸毒性(分別為250與16mg/Kg/天,按mg/m推算分別相當於人最大推薦劑量的800與100倍)。在大鼠中,在出現母體毒性的劑量125、150mg/Kg/天時,可見胎仔重量輕微降低,發育遲緩,但劑量為62.5mg/Kg/天(按mg/m推算相當於人最大推薦劑量的200倍)時未見改變。
在圍產期毒性試驗中,大鼠在妊娠與哺乳期經口給予右佐匹克隆達180mg/Kg/天。可見各劑量組著床后丟失增加,幼仔體重與存活率降低,幼仔驚嚇反應增強。最低劑量為60mg/Kg/天,按mg/m推算分別相當於人最大推薦劑量的200倍。試驗中未見明顯的母體毒性,對於代其他行為指標或生殖功能來見影響。
致癌性
SD大鼠經口給予右佐匹克隆的致癌性試驗中束見腫瘤發生率增加,量高劑量為16mg/Kg/天,血漿水平(AUC)估測為人量大推薦劑量下血漿水平的80倍(雌性動物)和20倍(雄性動物)。但在SD大鼠摻食法給予消旋佐匹克隆的致癌性試驗中,在劑量為100mg/Kg/天時,右佐匹克隆血漿水平高於上述右佐匹克隆致癌性試驗中所達到的血漿水平,可見雌性動物乳腺癌、雄性動物甲狀腺泡膜細胞腺瘤與癌發生率增加,此時,右佐匹克隆的血漿水平估測為人最大推薦劑量下水平的150倍(雌性動物)和70倍(雌性動物)。乳腺癌的發生機理尚不清楚。甲狀腺腫瘤的發生率增加,認為是循環中甲狀腺激素代謝增加縫發TSH水平升高所致,該機制認為與人類無相關性。
在B6C3F1小鼠致癌性試驗中,摻食法給予消旋佐匹克隆,在最高劑量100mg/Kg/天時,可見雌性動物肺臟腫瘤發生率增加,雄性動物皮膚纖維瘤與內瘤發生率增加。上述劑量下右佐匹克隆的血漿水平為人最高推薦劑量下水平的8倍(雌性動物)和20倍(雄性動物)。皮膚腫瘤的發生是由於動物攻擊行為所致,與人類無相關性,在一項CD-1小鼠的致癌性試驗中,經口給予右佐匹克隆達100mg/Kg/天,未見肺臟與皮膚腫癌發生率增加。該試驗中最高劑量下血漿水平估測為人最大推薦劑量下水平的90倍.即達到了上述消旋體試驗中暴露量的12倍。在P53轉基因小鼠試驗中,在經口給藥劑量達到300mg/Kg/天時,未見腫瘤發生率增加。
2.藥理作用:
右佐匹克隆是一種非苯二氮卓類催眠葯,右佐匹克隆催眠作用的確切機制尚不清楚,但認為是作用於與苯二氨卓受體偶聯的GABA受體複合物引起的。
對健康志願者(成人及老人)及肝腎疾病者進行了葯代動力學研究,健康受試者中,單劑量最高劑量達到7.5mg,並且進行了7天連續給葯試驗,劑量分別為1,3和6mg,本品可被快速吸收,大約1小時達峰(tmax),終相半衰期大約為6小時,健康成人連續服用本品不蓄積,在1-6mg;分佈與和量呈線性關係。
吸收與分佈
口服后本品快速吸收,口服后大約1小時達到血漿濃度峰值。血漿蛋白結合率低,為52%-59%。紅細胞非選擇性吸收。
代謝
口服后,本品主要通過氧化與去甲基化代謝,主要血漿代謝物為N-氧化右佐匹克隆與N-去甲基右佐匹克隆。N-去甲基右佐匹克隆與GABA受體結合率遠低於右佐匹克隆,N-氧化右佐匹克隆與GABA受體結合不緊密。體外實驗顯示右佐匹克隆代謝與CYP3A4與CYP2E1相關。右佐匹克隆對低溫儲藏肝細胞不顯示任何對CYP450 1A2,2A6,2C9,2C19,2D6,2E1,3A4的抑制作用。
消除
口服吸收后,右佐匹克隆消除半衰期大約為6小時,口服消旋佐匹克隆,劑量的75%以代謝物的形式在尿液中排出。右佐匹克隆的消除與佐匹克隆相似,小於10%口服劑量的右佐匹克隆以原形藥物從尿液中消除。
食物
健康成人,服用高膳食物后口服3mg右佐匹克隆,AUC未發生變化,平均Cmax降低21%,Tmax延遲1小時。半衰期未發生變化,大約為6小時。若在高脂/過多食物后立即服用右佐匹克隆,右佐匹克隆對睡眠潛伏期的作用可能降低。
特殊人群用藥
年齡
與成年人相比,65歲以上的患者AUC增加41%,半衰期大約為9小時,Cmax未發生明顯變化。
性別
男性與女性的葯代動力學參數相似。
種族
對I期臨床所有受試者數據進行分析,所有人種葯代動力學結果相似。
肝損傷
162健康志願者及8名患有輕度、中度、重度肝病患者進行了服用2mg右佐匹克隆的葯代動力學研究。嚴重肝損傷患者與健康志願者相比,暴露量增加了2倍,Cmax與tmax束髮生化。嚴重肝損傷患者服用的最高劑量應為2mg。輕度至中度肝損傷患者沒必要進行劑量調整。肝損傷患者服用右佐匹克隆應注意(見【用法用量】項)。
腎損傷
24名輕度、中度或重度腎損傷患者進行了葯代動力學研究。與健康對照者相比,AUC與Cmax相似。由於口服右佐匹克隆僅有小於10%通過尿液代謝,因此腎損傷患者沒必要進行劑量調整。
右佐匹克隆
基本信息
- 中文名
- 右佐匹克隆
- 外文名
- eszopiclone
- 別名
- 4-甲基-1-哌嗪甲酸6-(5-氯-2-吡啶基)-6
- 拼音
- You Zuo Pi Ke Long Pian
- 熔點
- 202 至 204 ℃
- 密度
- 1.54g/cm3
- 沸點
- 580.7°C at 760 mmHg
- 分子式
- C17H17ClN6O3
- 閃點
- 305 ºC
- EINECS號
- 202-303-5
- 分子量
- 388.8083
- 適應症
- 用於治療失眠
- CAS登錄號
- 138729-47-2
- 英文別名
- [(7S)-6-(5-chloropyridin-2-yl)-5-oxo-7H-pyrrolo[3,4-b]pyrazin-7-yl] 4-methylpiperazine-1-carboxylate;Lunesta;Esopiclone;Estorra;UNII-UZX80K71OE;
- 精確質量
- 388.80800
- PSA
- 91.76000
- 蒸汽壓
- 1.78E-13mmHg at 25°C
- 熔點1
- 202-204ºC
- 外觀與性狀
- 白色或略淡黃色粉末
- 成份
- 本品的活性成份為右佐匹克隆。
- 安全性描述
- S26;S36
- 危險性符號
- Xn
- 危險性描述
- R20/21/22;R36/37/38;R62
- 折射率
- 1.688
- 規格
- 2mg、3mg
- 性狀
- 本品為白色薄膜衣片,除去薄膜衣后顯白色或類白色。
- 商品名稱