組織細胞增多症
組織細胞增多症
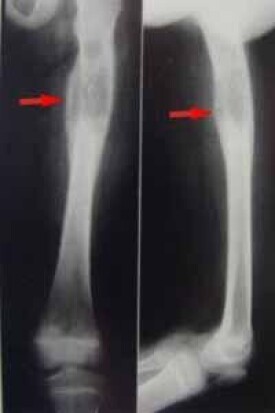
組織細胞增多症是一組單核-巨噬細胞(組織細胞)異常增生的疾病,較罕見,其中有Hand-Schüler-Christian病、Letter-Siwes病和嗜酸細胞肉芽腫病。因病因均未明,故定名為組織細胞增生症X。雖然各病種累及的臟器範圍(骨、皮膚、淋巴結、腦垂體等)、發病年齡、臨床表現各不相同。但肺都有不同程度累及,組織學改變也同樣為單核-巨噬細胞異常增生和嗜酸粒細胞浸潤而形成間質性肉芽腫。目前發現最多是惡性組織細胞病,故以惡性組織細胞病為代表作為本組闡述。
惡性組織細胞病(malignanthistiocytosiv)簡稱惡組,是組織細胞系的一種惡性增生性疾病。多見於成人和青壯年,但小兒時期也可發生少數病例。其主要的病理特點是肝、脾、淋巴結、骨髓等器官、組織中出現廣泛的惡性組織細胞或分化較高的組織細胞灶性浸潤,並伴有明顯的血細胞被吞噬現象。
組織細胞增多症
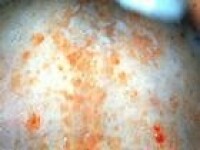
任何年齡均可患病,以15~40歲佔多數,男女之比約為3∶1,以農民為多見。臨床表現可分為急性型和慢性型(病程在1年以上)兩種,以急性型為多。起病急驟,病程短促、兇險,發熱為首見及常見的癥狀(97.2%),多為持續高熱,少數為不規則發熱,而隨病程進展而漸升高。少數也可以乏力、上呼吸道感染或肝、脾、淋巴結腫大起病。總之病情進展迅速,短期內明顯消瘦,極度衰弱。由於骨髓被大量異常組織細胞浸潤及組織細胞有吞噬血細胞的作用,加上脾亢及毒素抑制等作用,大多數患者有全血細胞減少。
組織細胞增多症
1.腸型惡組好發於結腸或小腸,累及不同腸段,腸壁增厚使腸腔狹窄,也可瀰漫浸潤,侵及整個腸壁。臨床有腸出血、壞死或潰瘍表現,穿孔率為88.8%。一旦穿孔,預后極差。
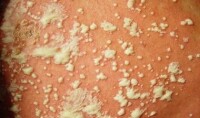
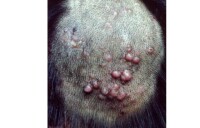
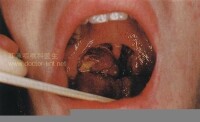
2.皮膚型惡組皮損為多形性,非向表皮性,多見於四肢,呈向心性分佈。特異性皮損表現為浸潤性斑塊、結節、潰瘍、剝脫性紅皮病或大皰等,呈進行性發展;個別自行消退或此起彼伏。本型起病緩慢,病程較長,呈慢性經過,高熱及肝脾腫大少見。
3.神經型惡組多累及9、10、12對顱神經麻痹,引起吞咽困難、眼球麻痹、失明。也可累及脊髓或表現為腦炎型。
4.多漿膜炎型多在全身浸潤,疾病進展期發生。漿膜腔積液2/3為血性,1/3為黃色漿液纖維素性,療效及預后極差。
組織細胞增多症
(二)骨髓檢查多數增生活躍,增生低下病例多已達晚期。多數病例骨髓中能找到多少不一的異常組織細胞:
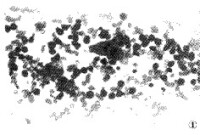
①異形組織細胞,細胞體積大,可達20~40μm,形狀畸異,核卵圓或不規則,核膜較厚而清晰,核染色質粗網狀,核仁隱顯不一,有時較大,胞漿嗜鹼性,常見空泡,可有纖細的嗜亞尼林藍顆粒;
②多核巨組織細胞:體積甚大,外形不規則,胞漿淺藍,無顆粒或僅有少數細顆粒,通常含核3~6個,彼此貼近或呈分葉狀,核仁隱顯不一,此類細胞較少見,在多數患者的骨髓塗片中不易找到,但在組織切片中易發現;
③吞噬型組織細胞:其形態與正常所見巨噬細胞類同,漿內常吞噬大量血細胞,包括幼紅細胞、成熟紅細胞殘片、血小板,偶有少數中幼粒細胞。此外,尚可見淋巴樣、單核樣、早幼粒樣、漿細胞樣及異常核絲分裂型組織細胞,其中異形組織細胞和(或)多核巨組織細胞對惡組有診斷意義。吞噬型、淋巴樣和單核樣組織細胞在其它疾病中都可出現,因此沒有特異性診斷價值。由於骨髓受累程度不一致,病灶分佈不均勻,因此一次骨髓穿刺陰性者,不能排除診斷,反覆多部位穿刺可提高診斷的陽性率。骨髓活檢或骨髓液凝塊檢查也可提高本病診斷陽性率。
組織細胞增多症
(四)位相顯微鏡及電鏡檢查位相顯微鏡下,細胞邊緣呈花邊樣,有"頭髮樣"胞漿突起,常有明顯的伸縮活動,核色質纖細,線粒體滿佈於胞漿內,有彙集於核凹陷部傾向,核膜清楚。惡組細胞的電鏡特徵是具有不規則短索狀的糙面內織網,線粒體小,胞質內有各種不同顆粒,分化好的常有吞噬物質。
(五)病理學診斷淋巴結穿刺、活檢加印片,有時需反覆多次、多部位檢查可提高診斷率。鏡下見淋巴結包膜下、髓索及淋巴實質內組織細胞增殖、浸潤、形態畸形,有吞噬性,常有少量漿細胞,但包膜多不侵犯。脾、肝、腎或皮膚活檢亦見類似的病變。
(六)染色體檢查惡組病的染色體核型變化常以多倍體為著,有較高比例的亞二倍體和超二倍體;亞二倍體染色體數為45條,且恆定特徵改變系D組染色體丟失一條,此與白血病、淋巴瘤不同,有助診斷。此外,可有染色體易位t(8;16)(p11;p13)及標記染色體(M)出現。
組織細胞增多症
組織細胞增多症
本病尚無理想治療方法,個別患者可適用單一化療藥物,環磷醯胺可作為首選,100~400mg/d口服或靜注,總量可達8~12g。緩解后可50~150mg/d維持。然多數患者對單一化療藥物幾乎均有抗藥,疾病兇險,須採用類似大細胞性非何傑金淋巴瘤聯合化療方案,自1977年Alexander採用聯合強力化療后療效有所改觀。
化療方案多採用:①CHOP:環磷醯胺750mg/m2,靜注,第1天;阿黴素50mg/m2,靜注,第1天,長春新鹼2mg,靜注,第1天;潑尼松50mg/(m2·d),口服,第1~5天。②B-CHOP:加用博來黴素20mg/m2,靜注,第1天。③B-CHOP-HDMTX,上述方案再加用大劑量氨甲喋呤0.25~20g/m2,靜脈滴注,24小時后給4氫葉酸鈣10mg/m2q6h×10次解救。Alexander(1984)採用聯合化療交叉方案CHOP/BCHOP及CHOP/B-CHOP±HDMTX治療22例,15例(68%)取得完全緩解,5例(23%)部分緩解,2例未緩解,中數緩解時間30月(9-105月),中數生存期2年,5年生存率40%。Vera(1984)首先採用依託泊甙(VP-16-213)+阿糖胞苷治療一例CHOP±HDMTX無效的惡組患者取得成功,無病生存、完全緩解期32月。劑量VP-16-21360mg/m2、阿糖胞苷200mg/m2靜脈滴注,每周一次×6周;以後VP-16-21340mg/m2、阿糖胞苷200mg/m2,每周一次×3周,此後隔周一次持續10月。
| 腦垂體 | 阿糖胞苷 | 霍奇金氏淋巴瘤 | 阿黴素 |
| 巨噬細胞 | 嗜亞尼林藍顆粒 | 血細胞 | 長春新鹼 |
| 非何傑金淋巴瘤 | 奔馬律 | 漿膜腔 | 重型再生障礙性貧血 |
1,http://www.cqvip.com/QK/96202X/2006004/22896044.html
2,http://www.wiki.cn/wiki/%E7%BB%84%E7%BB%87%E7%BB%86%E8%83%9E%E5%A2%9E%E5%A4%9A%E7%97%87
3,http://www.51qe.cn/pic/40/13/11/16/029.htm
4,http://www.yongyao.net/jbhtml/zzxbzdzx.htm