進行性肌營養不良症
由於基因缺陷所導致的肌肉變性病
進行性肌營養不良是一類由於基因缺陷所導致的肌肉變性病,以進行性加重的肌肉無力和萎縮為主要臨床表現。由於基因缺陷的不同,臨床癥狀出現的早晚不同,可以早至胎兒期,也可以在成年後。從疾病名稱就可以知道,肌營養不良的病程一般是進行性加重的,但疾病進展的速度快慢不一。
病理上以骨骼肌纖維變性、壞死為主要特點,臨床上以緩慢進行性發展的肌肉萎縮、肌無力為主要表現,部分類型還可累及心臟、骨骼系統。傳統上分為假肥大型肌營養不良、面肩肱型肌營養不良、肢帶型肌營養不良、Emery-Dreifuss肌營養不良、眼咽型肌營養不良、眼型肌營養不良、遠端型肌營養不良和先天性肌營養不良。按照遺傳方式可分為連鎖隱性遺傳型、常染色體顯性遺傳型和常染色體隱性遺傳型。
徠2018年5月11日,國家衛生健康委員會等5部門聯合制定了《第一批罕見病目錄》,進行性肌營養不良被收錄其中。
進行性肌營養不良症
假肥大型肌
最多見。現在亦被稱為抗肌萎縮蛋白缺陷型肌營養不良,又分為Duchenne型(Duchenne muscular dystrophy, DMD)和Becker型(Becker muscular dystrophy, BMD),前者發病率約為1/3500活產男嬰,後者發病率較低,約為1/20,000。。其他因抗肌萎縮蛋白缺陷引起的肌病包括X連鎖擴張型心肌病、肌痛肌痙攣綜合征、女性肌營養不良症等。
肢帶型肌營養不良
肢帶型肌營養不良(limb girdle muscular dystrophy, LGMD)是一組臨床表現和遺傳特點不同的異質性肌病,分為常染色體顯性遺傳和常染色體隱性遺傳兩大類型,稱為LGMD1和LGMD2,在此基礎上根據致病基因和缺陷蛋白又分為若干亞型,分別命名為LGMD1A、1B、1C和2A、2B、2C…等。
面肩肱型肌
面肩肱型肌營養不良(facioscapulohumeral musculardystrophy, FSHD)為常染色體顯性遺傳,發病率約為1~5/10~20萬。
Emery-Dreifuss肌
主要是性連鎖隱性遺傳,少數可為常染色體顯性或隱性遺傳,分別稱為EDMD1、EDMD2和EDMD3型。
遠端型肌營養不良
根據遺傳方式、基因定位、臨床上是以手肌、脛前肌為主,還是腓腸肌為主,將遠端型肌營養不良又分為多個亞型,在40歲前起病的Welander型、Markesberry-Grigg-Udd型,在40歲以後起病的Nonaka型、Miyoshi型和Laing型。
眼咽型肌營養不良
眼咽型肌營養不良(oculopharyngeal muscular dystrophy, OPMD),較少見,常染色體顯性或隱性遺傳,或為散發。
先天型肌營養不良
先天型肌營養不良(congenital muscular dystrophy, CMD)根據臨床表現、基因和生化缺陷被分為10多個類型。
進行性肌營養不良症是一組遺傳性疾病,多數有家族史,散發病例可為基因突變。在肌細胞膜外基質、跨膜區、細胞膜內面以及細胞核膜上有許多蛋白,基因變異可導致編碼蛋白的缺陷,導致肌營養不良。由於不同的蛋白在肌細胞結構中所起的作用不完全相同,導致不同類型的肌營養不良。
現代分子遺傳學發現肌營養不良與肌膜蛋白、近膜蛋白、核膜蛋白的缺陷有關。但蛋白缺陷如何引起肌肉變性壞死,導致肌肉進行性萎縮的機制仍不清楚。
假肥大型肌
包括DMD和BMD,是X連鎖隱性遺傳性疾病,致病基因為dystrophin,位於染色體Xp21,該基因是目前人類發現的最大的基因,長度約2400~3000kb,約79個外顯子,編碼3685個氨基酸,組成dystrophin蛋白(即抗肌萎縮蛋白),分子量427KD。Dystrophin蛋白位於骨骼肌和心肌細胞膜內面,為細胞骨架蛋白,具有抗機械牽拉作用,能防止肌細胞在收縮過程中的損傷。Dystrophin與細胞膜內面、跨細胞膜區以及細胞膜外區的多種蛋白如sarcoglycan、dystroglycan等蛋白緊密結合,相互關聯,在細胞膜內外組成一個整體,維繫細胞膜內外的物質交換和聯繫,保護細胞膜結構完整和穩定。Dystrophin基因缺陷導致肌細胞膜上dystrophin蛋白缺乏或減少,使肌細胞膜不穩定而引起肌細胞壞死和功能喪失。如dystrophin蛋白完全缺乏,產生DMD表現;如僅為量的減少,則為BMD。
肢帶型肌營養不良
LGMD主要與一大組肌膜蛋白和近膜蛋白的缺陷有關,如α、β、γ、δ-肌聚糖(sarcoglycan)之間相互連結,組成跨肌膜複合體,並與β-dystroglycan和dystrophin相互作用。基因突變導致相應肌聚糖亞單位不正確表達或不適當裝配,影響肌膜的穩定性,產生LGMD2D、2E、2C、2F。LGMD2A和2B的缺陷蛋白分別為calpain3和dysferlin。LGMD1A、1B、1C的缺陷蛋白分別為myotilin、lamininα2和caveolin3。
面肩肱型肌
FSHD基因定位於染色體4q35,其致病基因尚未克隆,基因產物尚未分離出來。FSHD為常染色體顯性遺傳,具有幾乎完全的外顯性,幾乎所有的FSHD患者都在4q35區域存在3.3kb重複片段的缺失,正常人該片段重複11~150次,而FSHD通常少於11次。這種基因重複片段的缺失並不直接破壞任何可識別的基因,而是使染色體端粒更接近著絲點,間接的增加相鄰基因的表達。在細胞核內染色質不適當的相互作用可能是致病原因之一,但確切發病機制仍不清楚。
Emery-Dreifuss肌
EDMD1型基因定位於Xq28,編碼762bp的mRNA。其34KD的蛋白產物由254個氨基酸構成,稱為emerin。emerin是錨定於骨骼肌、心肌和平滑肌核膜內表面的核被膜蛋白,其主要功能是在肌肉收縮過程中對抗機械性壓力以穩定核膜。目前已發現該基因突變形式包括點突變、小片段缺失和無義突變等。EDMD2和3型的基因為LMNA,定位於Iq11-23,編碼核纖層蛋白laminA/C。lamin是核膜的組成部分,定位於核膜板層,它在DNA複製、染色體構建、核孔複合體的空間構形、細胞核發育、核膜蛋白錨定等方面起作用。
眼咽型肌營養不良
OPMD基因為多聚腺核苷酸結合蛋白核1基因,定位於14q11.2-13。基因長2.4Mbp,突變發生於該基因的第一個外顯子上,由於基因突變使染色體在減數分裂和有絲分裂期異常擴增了GCG三核苷酸。
假肥大型肌
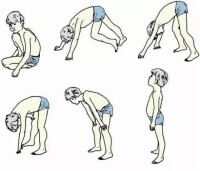
患兒運動發育較正常兒童晚,如學會走路晚、步態蹣跚、不能跑步、常無故摔倒。在3~5歲時癥狀逐漸明顯,因骨盆帶肌力弱,不能跳躍、奔跑,上樓費力,行走姿勢異常,腰椎過度前突,骨盆向兩側擺動,呈典型的“鴨步”。由於腹直肌和髂腰肌無力,患者由仰卧位起立時,先翻身轉為俯卧位,然後伸直雙臂用雙手支撐床面,雙腿亦伸直,逐漸用雙手扶住膝部,依次向上攀附大腿部,直到立起,這一動作是DMD的特有表現,稱為Gower征。萎縮無力肌肉開始主要是大腿和骨盆帶肌,逐漸發展至小腿肌、上肢近端、上肢遠端肌肉,最後呼吸肌麻痹。腓腸肌肥大常非常顯著,其他可出現舌肌、三角肌、臀肌等肌肉肥大。DMD常伴有心肌損害,累及心室、心房、傳導系統。晚期出現心臟擴大徠、心力衰竭,約10%患者可因心功能不全死亡。此外可出現關節攣縮、足下垂、脊柱側彎等。多數在12歲左右不能行走,20歲左右因呼吸肌無力、呼吸道感染,引起呼吸肌衰竭死亡。
BMD臨床表現與DMD類似,但發病年齡較晚,約為5~15歲,病情較輕,進展速度較慢,12歲以後仍能行走,,存活時間較長,部分可接近正常壽命。
肢帶型肌營養不良
常染色體隱性遺傳型較常見,發病較早,癥狀較重,在兒童、青春期或成年時起病,表現為骨盆帶肌和肩胛帶肌的肌肉萎縮無力,以致患者上樓費力,蹲起困難,雙上肢上舉困難,出現翼狀肩胛,面肌一般不受累。可有腓腸肌肥大。部分患者心臟受累。
面肩肱型肌營養不良
面肌力弱是首發癥狀,但因發病隱襲,癥狀較輕,常被忽略。表現為閉眼無力或閉眼露白,示齒時鼻唇溝變淺,不能吹口哨、鼓腮,嘴唇增厚而外翹,呈現典型的肌病面容。肩胛帶肌力弱,出現翼狀肩胛。胸大肌力弱,胸部萎陷。上肢近端、下肢近端和遠端肌肉均可受累。可見三角肌等肌肉肥大。部分病例合併滲出性視網膜炎和神經性聽力下降。
Emery-Dreifuss肌
5歲前起病,受累肌肉呈肱腓型,上肢以肱二頭肌和肱三頭肌為主,下肢則以腓骨肌和脛前肌,後期累及肩胛肌、胸帶肌及骨盆帶肌。肌無力或輕或重,沒有腓腸肌肥大。該病最主要特點是早期出現嚴重的關節攣縮,累及頸椎、肘、踝、腰椎等關節,使患者出現特殊的行走姿勢。另一個特點是心臟受累早,表現嚴重的傳導阻滯,心動過緩,心房纖顫,需要安裝起搏器。疾病緩慢進展,常因心臟病死亡。
眼咽型肌營養不良
起病年齡40~60歲,主要癥狀為雙側上瞼下垂,通常為對稱性,部分患者有不全性眼肌麻痹。咽喉肌力弱,吞咽困難,構音障礙。面肌、顳肌、咀嚼肌也可有輕的力弱。病情進展緩慢,但可因吞咽困難致營養不良或吸入性肺炎死亡。
遠端型肌營養不良
又稱遠端型肌病,表現為上肢或下肢遠端肌肉首先出現肌肉萎縮無力,特別是雙側手肌,下肢脛前肌和腓腸肌。根據遺傳方式、基因定位和受累肌肉不同分為若干亞型。
先天性肌營養不良
是一組先天性或嬰兒期起病的肌肉疾病,表現為肌張力低下、運動發育遲滯,可有進行性或非進行性肌肉萎縮、力弱,合併嚴重的骨關節攣縮和關節畸形,有腦和眼多系統受累,肌肉病理為肌營養不良改變。
根據典型病史、遺傳方式、陽性家族史、肌肉萎縮無力分佈特點,結合血清肌酶升高,肌電圖呈肌源性改變,肌肉活檢病理為肌營養不良或肌源性改變的特徵,多數肌營養不良症可獲得臨床診斷。進一步確診或具體分型診斷需要用抗缺陷蛋白的特異性抗體進行肌肉組織免疫組化染色以及基因分析。
(1)血清肌酶檢驗:包括肌酸激酶、乳酸脫氫酶、肌酸激酶同工酶、天冬氨酸氨基轉移酶和丙氨酸氨基轉移酶等。DMD時肌酸激酶升高顯著,可達正常值的20~100倍以上。BMD時可升高5~20倍。在疾病的不同階段,肌酶水平也有變化。早期升高顯著,當肌肉萎縮嚴重達疾病晚期時肌酶水平逐漸下降。LGMD和遠端型肌病患者肌酶輕到中度升高,FSHD患者肌酶可正常或輕度增高。
(2)肌電圖:肌電圖呈現典型肌源性改變的特徵,輕收縮時運動單位電位時限縮短,波幅降低,最大用力收縮時為電位密集的病理干擾相。在疾病不同階段,肌電圖改變也可有變化。
(3)肌肉活檢病理:肌營養不良肌肉組織病理表現為肌纖維變性、壞死,可見不透明纖維和肌纖維再生,可見肌纖維肥大,間質中結締組織和脂肪組織增生。DMD不同階段病理改變也不相同,在疾病晚期以結締組織增生為主,在大量結締組織中可殘存少數變性肌纖維。BMD的病理改變較DMD輕。LGMD可出現可出現分裂纖維和渦狀纖維。採用針對缺陷蛋白的特異性抗體進行肌肉組織的免疫組化染色,是目前鑒別各型肌營養不良症的主要方法。
(4)基因檢查:部分肌營養不良症可採用基因檢查獲得診斷,主要是DMD和BMD患者,有助於基因攜帶者檢出和產前診斷。運用多重PCR技術,能檢測dystrophin基因缺失和基因重複,對於非缺失型的突變不能檢出,對點突變可採用mRNA分析進行檢測。應用p13E-11標記的4q35EcoR1/Bln1雙重消化可檢測限制性片段長度,對FSHD進行基因診斷。對於LGMD來說,由於涉及的基因多,每種亞型的基因突變缺乏熱點,因此直接的基因檢查比較困難,應先根據免疫組織化學結果初步分型然後再進行DNA檢測。
(5)其他檢查:胸片、心電圖和超聲心動圖檢查可了解患者心臟受累情況。骨和關節X線可了解骨關節畸形。肺功能檢查有助於判斷疾病的嚴重程度。
(1)進行性脊髓性肌萎縮:主要是與少年型脊肌萎縮症(Kugelberg-Welander病)鑒別,該病表現為下肢近端力弱,站立時腹部前凸,行走時似鴨步,與DMD臨床表現相似。但肌電圖呈典型的神經源性改變,血清CK正常或輕度增高,肌肉活檢病理為神經源性損害有助於鑒別。
(2)酸性麥芽糖酶缺陷病:即糖原累積病Ⅱ型,其兒童型以肢體近端肌肉無力為主要表現,個別患者有心臟擴大,甚至心衰,常見呼吸衰竭,於3~24歲死亡,與DMD相似。成人型表現為緩慢進展的進行性、對稱性四肢肌肉萎縮、力弱,近端比遠端重,軀幹肌及骨盆帶肌明顯,易誤診為LGMD。肌肉活檢病理檢查是主要的鑒別方法,酸性麥芽糖酶缺陷病可發現肌纖維空泡狀改變,PAS染色深染,分佈不均勻,酸性磷酸酶染色強陽性。電鏡下可見肌膜下、肌絲間糖原累積。肌肉組織、培養的成纖維細胞、淋巴細胞測定酸性麥芽糖酶減少可以確診。
(3)慢性多發性肌炎:成年人對稱性肢體近端無力,血清肌酶升高,是慢性多發性肌炎和LGMD的共同特徵,但前者沒有家族遺傳史,病情進展較快,多有肌痛,肌肉病理符合肌炎改變,用皮質類固醇激素或免疫抑製劑治療有效,不難鑒別。
(4)Charcot-Marrie-Tooth病:遠端型肌病主要表現為下肢遠端伸肌及屈肌萎縮力弱,因而與Charcot-Marrie-Tooth病(CMT)的臨床表現相似。但CMT的典型表現為大腿及前臂下1/3以下的肌肉萎縮,有或無感覺減退,肌電圖表現為神經源性損害,CMT1A運動傳導速度顯著減慢,腓腸神經病理有髓鞘脫失以及增生性的洋蔥球狀改變等可以鑒別。
(6)進行性眼外肌麻痹:易於眼咽型肌營養不良混淆。該病為一線粒體肌病,表現為上瞼下垂,眼球活動受限,可伴有四肢近端的肌無力。肌肉活檢病理在改良的Gomori三色染色下可見肌膜下出現不規則的紅色邊緣,即不整邊紅纖維(ragged red fiber, RRF),電鏡下證實為堆積的線粒體膜,進行線粒體DNA分析也有助於診斷。
進行性肌營養不良症是一大類基因突變引起的肌肉變性疾病,迄今尚無特效的治療方法。
皮質類固醇激素是目前唯一一個能夠在一定時間內保持DMD患者肌力的藥物。有6個雙盲試驗發表了最後的結果。目前多數採用,潑尼松O.75mg/kg/d,使用時間超過6個月,如出現副作用,如體重顯著增加,發育遲緩,骨質疏鬆等,則可將劑量減少至O.3mg/kg/d。也有採用潑尼松O.75mg/kg/d,每月前10天用藥,后20天不用的療法,認為可減輕副作用。另外,Deflazacort是潑尼松的衍生物,用於治療肌營養不良,無體重增加和骨質疏鬆的副作用,不良反應較潑尼松少。由於激素、免疫抑製劑並不能使肌纖維的dystrophin蛋白及其相關蛋白增多,並不能從根本上改變病程。
正常骨骼肌中有衛星細胞,在肌肉損傷後進行再生,分化形成新的肌細胞。當肌細胞在體外培養時,衛星細胞可發育成成肌細胞,將這些培養的大量的成肌細胞注入病變肌肉,使正常的成肌細胞與DMD的病肌細胞融合,達到治療目的,稱為成肌細胞移植。這種治療試驗研究已有20年之久,在DMD動物模型肌肉中出現了dystrophin陽性纖維的表達,但在試用於患者時,發現dystrophin陽性纖維非常少,臨床功能改善不理想,至今未能取得較好的效果。
已經進行包括骨髓幹細胞、血源性以及肌肉源性CD133抗原細胞、肌源性幹細胞、成血管細胞(mesoangioblast)、人源性周細胞(human derived pericyte)等幹細胞的移植試驗。在動物試驗中顯示出一些有希望的結果,可能為肌營養不良症的細胞治療提供新的思路。
目前仍處於探索階段。
適當鍛煉,合理營養,採取物理治療和矯形治療以糾正骨關節畸形,防治關節攣縮,對儘可能長地保持運動功能具有重要作用。加強呼吸鍛煉,改善呼吸功能和心臟功能,對防治呼吸和心力衰竭,較長時間維持生命有一定意義。進行心理治療,進行日常生活能力訓練,使患者和家庭保持積極的態度也非常重要。
DMD患者預后不良,在20歲左右死於呼吸衰竭或心力衰竭;LGMD的個別亞型和先天性肌營養不良症預后也較差;BMD、FSHD、眼咽型肌營養不良症和遠端型肌營養不良症預后相對較好,喪失運動功能的時間較晚,部分患者壽命可接近正常人。
進行性肌營養不良症是一組遺傳性肌病,其中假肥大型肌營養不良症癥狀嚴重,進展迅速,生命早期即喪失運動功能,且早期死亡,給家庭和社會造成很大負擔,而目前尚無特效的治療方法,因此早期檢出基因攜帶者,對其婚配、孕育進行指導,對胎兒進行產前診斷,早期人工流產高風險胎兒顯得非常重要。首先,應確定先症者的基因異常,然後採用基因技術檢查確定其母親是否為攜帶者,若為攜帶者,在懷孕以後應確定胎兒性別,若為男胎在應在妊娠8~17周時取羊水細胞或絨毛膜細胞,進行基因檢查,若高度懷疑為病胎,則應終止妊娠。對於常染色體隱性遺傳型肌營養不良,則應避免近親婚配。需要注意的是,雖然攜帶者檢出和產前診斷技術均有了發展,但仍存在許多問題,特別是涉及醫學倫理學和法律方面的問題,在實際臨床應用方面受到制約。
給予適當心理支持,使患者及家屬能面對現實,保持積極的心態,儘可能提高生活質量,延緩生存期限。
營養指導,給與高蛋白飲食,防止感冒、感染、褥瘡等疾病,監測患者肢體功能和心、肺功能。協助按摩、理療。在患者接受矯形手術時給與必要護理。
患者肌萎縮無力,起床、行走、洗簌、進食、如廁等既不方便,如何訓練和幫助患者適應疾病狀態下的生活非常重要。
基本信息
- 中文名
- 進行性肌營養不良症
- 外文名
- progressive myodystrophy
- 原因
- 遺傳
- 是否傳染病
- 否
- 是否遺傳
- 是
- 所屬科室
- 內科-神經內科