米氏方程
表示酶促反應起始速度的方程
米氏方程(Michaelis-Menten equation)表示一個酶促反應的起始速度與底物濃度關係的速度方程。
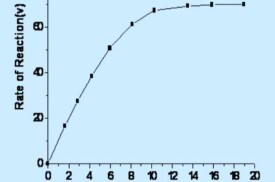
在酶促反應中,在低濃度底物情況下,反應相對於底物是一級反應(first order reaction);而當底物濃度處於中間範圍時,反應(相對於底物)是混合級反應(mixed order reaction)。當底物濃度增加時,反應由一級反應向零級反應(zero order reaction)過渡。
由此可見 值的物理意義為反應速度 達到 時的底物濃度(即),單位一般為,只由酶的性質決定,而與酶的濃度無關。可用 的值鑒別不同的酶。
當底物濃度非常大時,反應速度接近於一個恆定值。在曲線的這個區域,酶幾乎被底物飽和,反應相對於底物S是個零級反應。就是說再增加底物對反應速度沒有什麼影響。
米氏常數 是酶促反應速度n為最大酶促反應速度值一半時的底物濃度。這可通過用[S]取代米氏方程中的 證明,通過計算可得。
1913年Michaelis L.和Menten M.根據中間複合體學說提出了單底物酶促反應的 快速平衡模型或 平衡態模型(equilibrium-state model),也稱為 米-曼氏模型(Michaelis-Menten model):
式中E是酶,S是底物,ES是中間複合體,P是產物,是ES的 解離[平衡]常數,即第一步的逆向反應中的速率常數 和正向速率常數 之比,是催化常數,即第二步中的向前速率常數。
在建立模型和推導模型的速率方程時,他們實際上做了以下幾點假設:
①為了簡化起見,假設反應中只有一個中間複合體,反應的第一步 是可逆反應,並保持始終;
②反應的第二步 是限速步驟,這裡是限速步驟,這裡,也就是說ES分解生成P的速率不足以破壞E和ES之間的快速平衡;
③為了達到平衡,只用初始底物濃度 的很小一部分,因為一般情況下(初始酶濃度),因此在反映的初期,底物濃度[S]可以用 代替,或是把[S]看作;
④酶在反應中不被消耗,只是或以遊離形式E存在或以結合形式ES存在,因此遊離酶濃度[E]和中間複合體濃度[ES]只和等於初始酶濃度 或總酶濃度,即,這就是所謂的 酶守恆公式(conservation equation of enzyme);
⑤該模型沒有考慮 這一逆反應,但顯然 是一個不等於零的常數,要忽略這一步,必需使[P]接近於零,因此米-曼氏方程只適用於反應的初速率。
根據平衡態模型S轉變成P的總速率應由限速反應(模型中第二步)決定,因此產物生成速率
ES複合體的濃度[ES]在實驗上不易測定,需要找出容易測定的其他參數(如某些常數和已知的 等)來代替它。為此利用第一步反應(快速平衡)中ES解離成E和S的解離常數
則
將酶守恆公式 代入上式得
經整理得
代入 得
這裡 具有特殊的意義。當底物濃度[S]高至使所有酶分子都被飽和時,則,反應初速率 將達到最大值,用數學式可表示為
因此 也可寫成
平衡態模型中前兩點假設不具有普遍性,特別是沒有理由認為所有酶促反應的 都遠小於。因此1925年Briggs G. E.和Haldane J. B. S.對該模型提出了修正,但仍保留米-曼氏假設的后三點。他們用穩態模型(steady-state model)或稱 Briggs-Haldane氏模型:
代替了平衡態模型。對觀測初速率(即產物P尚未生成或很少生成時)來說,式中仍可忽略不計。所謂穩態是指反應進行不成的一段時間內(順便提及,幾毫秒內,這段時間的狀態稱為 前穩態),系統中[ES]由零增加到一定值,在一定時間內雖然[S]和[P]在不斷變化,ES複合體也在不斷地生成和分解,但ES的生成速率 與分解速率 接近相等,[ES]基本保持不變。因此在穩態下ES形成的凈速率,
因為 且
所以
整理得
這裡,速率是常數之比 本身也是一個常數,並被定義為 米氏常數(Michaelis constant), :
將 代入,整理得:
根據穩態模型,S轉變為P的速率決定於穩態濃度和限速的速率常數。因此
將 代入上式,得
或
根據兩種模型推導出的速率方程形式上是一樣的,兩者不同的是 比 具有更大的普遍性。穩態下,當 時,則,因此可以把平衡態看成是穩態的一個特例。為了紀念Michaelis和Menten兩人,人們把上述帶三角符號的的方程都稱為 米-曼氏方程(Michaelis-Menten equation)。
①當 時, 。因此,Km等於酶促反應速度達最大值一半時的底物濃度。
②當 時, 。因此,Km可以反映酶與底物親和力的大小,即值越小,則酶與底物的親和力越大;反之,則越小。
是酶的特徵性常數:在一定條件下,某種酶的Km值是恆定的,因而可以通過測定不同酶(特別是一組同工酶)的Km值,來判斷是否為不同的酶。
⑤ 可用來判斷酶的最適底物:當酶有幾種不同的底物存在時,Km值最小者,為該酶的最適底物。
⑥ 可用來確定酶活性測定時所需的底物濃度:當,為最合適的測定酶活性所需的底物濃度。
⑦ 可用於酶的轉換數的計算:當酶的總濃度和最大速度已知時,可計算出酶的轉換數,即單位時間內每個酶分子催化底物轉變為產物的分子數。
⑷ 和 的測定:主要採用Lineweaver-Burk雙倒數作圖法和Hanes作圖法。
酶促反應中的 和 值有幾種測量方法。固定反應中的酶濃度,然後分析幾種不同底物濃度下的起始速度,就可獲得 和 值。但直接從起始速度對底物濃度的圖中確定 或 值是很困難的,因為曲線接近 時是個漸進過程。所以通常都是利用米氏方程的轉換形式求出 和 值。常用的米氏方程轉換形式是Lineweaver-Burk方程,也稱為雙倒數方程。
使作圖,可以獲得一條直線。從直線與x軸的截距可以得到的絕對值;而是直線與y軸的截距。雙倒數作圖直觀、容易理解,為酶抑制研究提供了易於識別的圖形。
缺點:底物濃度低時,坐標點集中於坐標左下方,使得誤差增大,往往偏離直線,無法精確定出。
解決方法:底物濃度配成的濃度級差,而不是[S]的濃度極差,使點距離平均,再以最小二乘法線性回歸分析。
1、底物濃度對酶促反應速度的影響
當底物濃度很低時,有多餘的酶沒與底物結合,隨著底物濃度的增加,中間絡合物的濃度不斷增高。
當底物濃度較高時,液中的酶全部與底物結合成中間產物,雖增加底物濃度也不會有更多的中間產物生成。
2、溫度對酶反應速度的影響
一方面是溫度升高,酶促反應速度加快。另一方面,溫度升高,酶的高級結構將發生變化或變性,導致酶活性降低甚至喪失。因此大多數酶都有一個最適溫度。在最適溫度條件下,反應速度最大。
3、pH值對酶反應速度的影響
酶促反應速度受介質pH的影響,一種酶在幾種pH介質中測其活力,可看到在某一pH時酶促效率最高,這個pH稱為該酶的最適pH。酶作用存在最適pH提示酶分子活性基團的電離狀態、底物分子及輔酶與輔基的電離狀態都與酶的催化作用相關,但酶的最適pH也不是酶的特徵性常數,如緩衝液的種類與濃度,底物濃度等均可改變酶作用的最適pH。
4、激活劑對酶反應速度的影響
凡能提高酶活性的物質,都稱為激活劑。
(2)中等大小的有機分子:某些還原劑、乙二胺四乙酸(EDTA)
5、抑製劑對酶作用的影響
(1)不可逆抑制作用:抑製劑與酶的結合(共價鍵)是不可逆反應,抑製劑與酶結合后不能用透析等方法除去抑製劑而恢復酶活性。如二異丙基氟磷酸對胰凝乳蛋白酶或乙醯膽鹼酯酶;碘乙酸、碘乙醯胺、對一氯汞苯甲酸對巰基酶。
競爭性抑制作用是指有些抑製劑和底物結構極為相似,可和底物競爭與酶結合,當抑製劑與酶結合后,就妨礙了底物與酶的結合,減少了酶的作用機會,因而降低了酶的活力。它的特徵是,當加大底物濃度時,底物和酶結合的幾率增大,減少了抑製劑與酶的結合,抑制作用便會減弱。
非競爭性抑制作用是指有些抑製劑和底物可同時結合在酶的不同部位上,即抑製劑與酶結合后,不妨礙再與底物結合,但所形成的酶一底物一抑製劑三元複合物(ESI)不能發生反應。高濃度的底物不能使這種抑制作用逆轉。
值增大,值不變
值不變,值變小
值變小,值變小,但 值不變
基本信息
- 中文名
- 米氏方程
- 外文名
- The Michaelis Menten equation
- 提出時間
- 1913年
- 研究對象
- 酶促反應
- 類型
- 速度方程
- 計算公式
- v=Vmax×[S]/(Km+[S])
- 別名
- 米-曼式方程
- 提出者
- Michaelis L.、Menten M.
- 適用領域
- 化學