注射用奧馬珠單抗
一種重組的人源化單克隆抗體
注射用奧馬珠單抗是一種重組的人源化單克隆抗體,為抗IgE靶向生物製劑,是全球首個批准治療中至重度哮喘的靶向治療藥物,該產品於2003年首次在全球上市,已在96個國家獲得批准,包括美國、歐盟、日本等,已有超過680,000患者治療年的奧馬珠單抗累積暴露。已完成77個過敏性哮喘研究。在中國是治療中至重度哮喘患者的首個生物製劑,其療效和安全性明確,本品的上市,為我國中重度過敏性哮喘患者帶來更多個性化用藥的治療選擇。
注射用奧馬珠單抗
吸入性糖皮質激素(ICS)、短效(SABA)及長效β2-腎上腺素受體激動劑(LABA)、白三烯拮抗劑(LTRA)是臨床常用治療哮喘藥物,儘管大部分哮喘患者應用標準治療后癥狀明顯改善,但仍有約40%的哮喘患者在接受了全球哮喘防治倡議(GINA)指南所推薦的第4級療法后,其病情仍未得到充分控制,且因為哮喘急性發作導致住院或相關的死亡風險增高。如果長期口服糖皮質激素治療,這些哮喘患者將會產生多重副作用。
【通用名稱】注射用奧馬珠單抗
【英文名稱】Omalizumab for Injection
【漢語拼音】Zhu She Yong Ao Ma Zhu Dan Kang
使用本品后罕見報道過敏反應,表現為支氣管痙攣、低血壓、暈厥,蕁麻疹和/或喉嚨或舌頭的血管性水腫。過敏反應可發生於首次注射后,但也可在治療一年後發生。本品注射后需要在合適的時間內密切觀察患者,並做好處理嚴重過敏反應的準備。告知患者過敏反應的常見癥狀和體征,提醒出現相關癥狀應立即就醫。
活性成份為奧馬珠單抗。
奧馬珠單抗為採用基因重組技術以中國倉鼠卵巢細胞生產的人免疫球蛋白E人源化單克隆抗體。
分子結構:奧馬珠單抗由兩條450-或451個氨基酸殘基組成的重鏈和兩個218個氨基酸殘基組成的輕鏈構成。兩條重鏈都含有連接在蛋白骨架的Asn301低聚糖鏈。
分子量:約為150,000道爾頓。
凍乾粉輔料:蔗糖,L-組氨酸,L-鹽酸組氨酸一水合物和聚山梨酯20。
稀釋液:滅菌注射用水。
復溶后,每瓶中奧馬珠單抗濃度為125 mg/mL(150 mg溶於1.2 mL溶劑)。
凍干品為白色至類白色塊狀疏鬆體,復溶後為澄清至乳光液體。
注射用奧馬珠單抗僅適用於治療確診為IgE(免疫球蛋白E)介導的哮喘患者(見【用法用量】)。
本品適用於成人和青少年(12歲及以上)患者,用於經吸入型糖皮質激素和長效吸入型β-腎上腺素受體激動劑治療后,仍不能有效控制癥狀的中至重度持續性過敏性哮喘。本品能降低這些患者的哮喘加重率。
150mg/瓶
本品應由具有診斷和治療中至重度持續性哮喘經驗的醫生使用。
用量
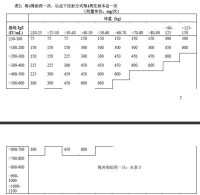
根據基線IgE(IU/mL,治療開始前測定)和體重(kg),確定本品合適的給藥劑量和給葯頻率。開始給葯前,應採用市售血清總IgE測定產品檢測患者IgE水平,以確定給藥劑量。
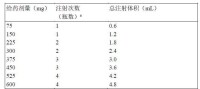
根據上述測定結果,每次給藥劑量為75 - 600 mg,按照需要分1 - 4次注射。
IgE水平低於76 IU/ml的患者獲益不明顯。對於IgE水平低於76 IU/ml的成人和青少年,在開始治療前,處方醫生應確認體外測定(RAST)結果已明確其對常年性過敏原過敏。
表1為換算表,表2和表3為成人和青少年(12歲及以上)的劑量確定表。
基線IgE水平或體重(kg)在給藥劑量表範圍外的患者,不應給予本品治療。
本品最大推薦給藥劑量為600 mg,每2周給葯一次。
表1:每次給葯時,給藥劑量與瓶數、注射次數和總注射體積的換算
注射用奧馬珠單抗
注射用奧馬珠單抗
表3:每2周給葯一次,以皮下注射方式每2周注射本品一次
(劑量單位:mg/次)
注射用奧馬珠單抗
治療療程、監測和劑量調整
本品用於長期治療。臨床試驗證明,至少經過12-16周應用本品治療后,才能顯示出有效性。本品治療16周時,應由患者的主治醫師對患者的治療有效性進行評價,以確定是否繼續給葯。本品治療16周后或後續治療中,應根據總體哮喘控制效果是否出現顯著改善,決定是否繼續應用本品的治療。
中止本品治療通常會導致遊離IgE水平恢復至較高水平和相關癥狀的複發。治療期間總IgE水平升高,且治療中止一年內總IgE仍維持高水平。因此,不能根據本品治療期間重新測得的IgE水平重新確定本品的給藥劑量。治療中斷不足一年時,給藥劑量的確定應以首次劑量確定時測得的血清IgE水平為依據。只有當本品治療已經中斷一年或以上時,才可以根據重新測得的總血清IgE水平確定給藥劑量。
當體重發生顯著變化時,應調整劑量(見表2和3)。
用法
僅供皮下注射使用。不得採用靜脈注射或肌肉注射給藥方法。
在上臂的三角肌區進行皮下注射給葯。如果因一些原因不能在三角肌區注射,也可在大腿部注射給葯。
患者自行注射本品的經驗有限。所以,本品僅供醫療保健專業人員給葯。
使用和處理指導原則
凍乾產品需要15~20分鐘方可溶解,有時可能需要更長時間。完全復溶的產品應澄清或略顯不透明,可能在瓶的邊緣有少量氣泡或泡沫。因為復溶產品具有一定的粘度,所以在從注射器中排除空氣或過量溶液前,必須從瓶中小心取出全部產品,以得到1.2 mL注射液。
製備本品 150 mg瓶裝製劑的皮下注射液,請遵照下列要求操作:
1. 用配備大內徑(18號)針頭的注射器從安瓿瓶中抽取1.4 mL滅菌注射用水。
注射用奧馬珠單抗
2. 將瓶直立於平面上,使用標準無菌方法將針頭插入瓶中,將滅菌注射用水直接注射進裝有凍乾粉的瓶中。
注射用奧馬珠單抗
3. 保持瓶直立,用力旋動以使瓶中液體呈漩渦狀(不得振蕩)約1分鐘,均勻潤濕粉末。 4. 為了幫助溶解,在完成第3步后,約每5分鐘輕輕旋動瓶身5~10 秒,溶解所有殘留固體。
4. 為了幫助溶解,在完成第3步后,約每5分鐘輕輕旋動瓶身5~10 秒,溶解所有殘留固體。
注射用奧馬珠單抗
* 注意:有時可能需要20分鐘以上粉末才能完全溶解。如果出現這種情況,重複第4步,至溶液中無凝膠樣可見顆粒。
產品完全溶解時,溶液中應無凝膠樣可見顆粒。瓶邊緣有小氣泡或泡沫屬正常現象。復溶產品為澄清或略顯不透明溶液。如果溶液中存在固體顆粒,不得使用。
5. 將瓶倒置至少15 s,使溶液流向瓶塞處。使用配備大內徑18號針頭的新的3 mL注射器,將針頭插入倒置的瓶中。保持安瓿瓶倒置,注射器抽取溶液時,針尖置於瓶塞內溶液的最底端。從瓶中取出針頭前,將注射器內芯一直拉到注射器桶的末端,以保證從倒置瓶中取出所有溶液。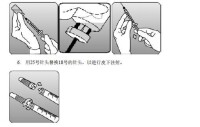
注射用奧馬珠單抗
6. 用25號針頭替換18號的針頭,以進行皮下注射。

注射用奧馬珠單抗
8. 在上臂的三角肌區域或大腿部進行皮下注射給葯,注意避開蕁麻疹病灶。
從微生物角度來說,本品復溶后應立即使用(參見【有效期】)。
按照當地法規要求,對於任何未使用的藥物或廢棄材料需要進行處理。
特殊人群
腎損害或肝損害
尚未研究肝腎功能損害對本品葯代動力學的影響。在臨床劑量水平,本品主要由網狀內皮系統(RES)清除,而不太可能受腎或肝損害影響。儘管無特別的劑量調整建議,在上述患者中應謹慎使用本品(參見【注意事項】)。
安全性特徵的總結
12歲和12歲以上成人和青少年患者臨床試驗期間,最常見不良反應為頭痛和注射部位不良反應,包括注射部位疼痛、腫脹、紅斑和瘙癢。6 至<12歲兒童臨床試驗中,最常見的不良反應為頭痛、發熱和上腹痛。這些反應多為輕度或中度。
不良反應列表
表4按照MedDRA系統器官分類和發生頻率列出了本品治療過敏性哮喘的安全性總人群在臨床研究中的不良反應。各發生率類型中不良反應按嚴重性遞減順序列出。發生頻率分類定義:十分常見(≥ 1/10),常見(≥ 1/100 至 < 1/10),偶見(≥1/l,000 至 < 1/100),罕見(≥ 1/10,000 至 < 1/1,000),十分罕見(< 1/10,000)。上市后報告的不良反應發生率類型列為未知(不能根據已有數據預估)。

注射用奧馬珠單抗
動脈血栓栓塞事件(ATE)
在對照臨床試驗和觀察性研究的中期分析中,發現ATE的數目存在不均衡性。複合終點ATE的定義包括卒中、短暫性腦缺血發作、心肌梗死、不穩定型心絞痛和心血管病因死亡(包括原因不明的死亡)。觀察性研究的終期分析顯示,在本品治療患者中,每1000患者年的ATE發生率為7.52(115/15,286患者年),對照組患者為5.12(51/9,963患者年)。在基線心血管風險因素多變數分析中,風險比為1.32(95%置信區間為0.91-1.91)。在另一項匯總的臨床試驗分析中,包括所有隨機、雙盲、安慰劑對照、為期8周或以上的臨床試驗,在應用本品治療患者中,每1000患者年的ATE發生率是2.69(5/1,856患者年),對照組為2.38(比率比1.13, 95%置信區間為0.24-5.71)。
血小板
臨床試驗中,少數患者的血小板計數低於正常實驗室範圍下限。這些變化均與出血發作或血紅蛋白減少無關。同非人類靈長類動物中(參見【藥理毒理】)結果相似,即使上市后報告了零散的特發性血小板減少症病例包括重度病例,在人類(6歲以上患者)中也未報告血小板計數持續性下降。
寄生蟲感染
高風險慢性蠕蟲感染的過敏患者中,安慰劑對照試驗結果表明,本品治療時感染髮生率略有增加,但無統計學顯著變化。感染病程、嚴重程度和治療應答均不變(參見【注意事項】)。
在中至重度哮喘患者和CSU患者的臨床試驗和上市后報告中有系統性紅斑狼瘡(SLE)病例的報道。但SLE的發病機理尚不明確。
嚴重過敏反應
在上市后報告中,基於約超過500,000患者年的暴露量中觀察到的嚴重過敏性反應總數,暴露於本品的患者嚴重過敏反應的發生頻率約為0.2%。
與本品無關的嚴重過敏反應病史可能是使用本品后出現嚴重過敏反應的一個風險因素。
惡性腫瘤
12歲和12歲以上成人和青少年的初始臨床試驗中,活性藥物治療組和對照組在腫瘤發病方面存在一些不均衡。活性藥物和對照組中發現的病例數為偶見(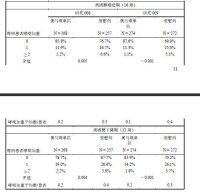 表6:研究008類固醇穩定期中哮喘癥狀和肺功能
表6:研究008類固醇穩定期中哮喘癥狀和肺功能 哮喘癥狀量表:總評分為0(最小)至9(最大);夜間和日間評分為0(最小)至4(最大癥狀)。
哮喘癥狀量表:總評分為0(最小)至9(最大);夜間和日間評分為0(最小)至4(最大癥狀)。
對本品活性成份或者其他任何輔料有過敏反應者禁用。
一般說明
本品不適用於急性哮喘加重、急性支氣管痙攣或哮喘持續狀態的治療。
本品尚未對自身免疫性疾病、免疫複合物介導疾病及已有腎損害或肝損害患者中進行治療的研究(參見【用法用量】)。該患者人群應慎用本品。
建議不要在開始本品治療后突然中斷全身或吸入型糖皮質激素治療。應在醫師的直接監督下減少糖皮質激素的用量,可逐漸降低劑量。
對操作人員的建議
僅可以採用皮下注射給藥方法,不得採用靜脈注射或肌肉注射給藥方法。
免疫系統疾病
· I型變態反應
本品治療時可能出現I型局部或全身變態反應(包括過敏性反應和過敏性休克),長期治療后也可能發生上述反應。大部分反應在第一次和後續注射本品的2小時內出現,但有一些反應發生在2小時以後,甚至發生在注射24小時后。因此,使用本品治療后,患者應始終有急救用治療過敏反應的藥品。應告知患者可能出現此類反應,如果發生過敏反應,應立即尋求醫療救助。
臨床試驗中極少出現過敏反應(參見【不良反應】)。
在少量臨床試驗患者中檢出了抗奧馬珠單抗的抗體(參見【不良反應】)。尚未完全了解抗奧馬珠單抗抗體的臨床相關性。
· 血清病
在人源化單克隆抗體(包括奧馬珠單抗)治療患者中,出現血清病和血清病樣反應(遲發的III型變態反應)。可能的病理生理學機制包括因出現抗奧馬珠單抗的抗體而導致免疫複合物的形成和沉積。典型發作時間為第一次,或後續注射給葯后,或在長期治療后1-5天。血清病癥狀包括關節炎/關節痛、皮疹(蕁麻疹或其它類型皮疹)、發熱和淋巴結病。抗組胺葯和糖皮質激素可用於預防和治療該疾病,應建議患者報告任何可疑癥狀。
· Churg-Strauss綜合征和嗜酸性粒細胞增多綜合征
重度哮喘患者很少出現全身性嗜酸性粒細胞增多綜合征或過敏性嗜酸性肉芽腫性血管炎(Churg-Strauss綜合征),常用全身性糖皮質激素治療上述癥狀。
在罕見情況下,抗哮喘藥物(包括奧馬珠單抗)治療患者存在或出現全身性嗜酸性粒細胞增多和血管炎。這些事件通常與口服糖皮質激素劑量下降有關。
在這些患者中,醫生應警惕患者出現顯著嗜酸性粒細胞增多、血管炎性皮疹、肺部癥狀加重、鼻旁竇異常、心臟併發症和/或神經病。
重度病例中出現上述免疫系統疾病時,應停止本品治療。
寄生蟲(蠕蟲)感染
IgE可能參與一些蠕蟲感染的免疫應答。在慢性高風險蠕蟲感染患者中,其過敏患者的安慰劑對照試驗結果表明,本品治療時感染治療的療程、嚴重程度和治療應答均無變化,但感染率略有增加。在全部臨床項目中沒有設計檢測該項疾病,且蠕蟲病的發生率低於1/1000。但是,蠕蟲感染高風險患者應謹慎用藥,特別是到蠕蟲感染盛行的地區旅行時。如果患者對推薦的抗蠕蟲治療沒有應答,應考慮停用本品。
對駕駛和操作機器能力的影響
本品對駕駛和操作機械的能力沒有影響或影響可以忽略不計。
有生育能力婦女
對於有生育能力婦女沒有特殊建議。
妊娠
關於孕婦應用本品的數據有限。動物研究表明,本品對生殖系統毒性均未產生直接或間接的有害作用(參見【藥理毒理】)。本品可通過胎盤屏障,尚不確定對胎兒是否有潛在傷害。非人類靈長類動物中,本品治療與年齡依賴的血小板計數下降有關,在幼年動物中對本品治療更為敏感(參見【藥理毒理】)。除非確實必須,否則妊娠期間不應使用本品。
哺乳
尚不明確本品是否分泌至人乳汁中。非人類靈長類動物的資料顯示本品可分泌至乳汁中(參見【藥理毒理】),不能排除對新生兒/嬰兒的風險。因此,在哺乳期間不應給予本品治療。
生育力
未獲得本品相關的人類生育力數據。在專門設計的非人類靈長類動物的非臨床生育力研究(包括交配研究)中,重複給予多達75 mg/kg的本品后,雄性和雌性動物的生育力均未發現損害。此外,在各項非臨床遺傳毒性研究中未發現遺傳毒性作用(參見【藥理毒理】)。
尚未明確本品在兒童人群(12歲以下)中應用的有效性和安全性。
尚未在6歲以下兒童患者中進行本品的臨床試驗。本品在6-11歲的兒童患者的臨床試驗主要在高加索人群中進行。
老年患者(65歲及以上)使用本品的數據有限,但無證據表明老年患者需要的劑量不同於65歲以下成人患者。
由於一些蠕蟲感染的免疫應答可能涉及IgE,本品可能間接降低治療蠕蟲或其他寄生蟲感染藥物的療效(參見【注意事項】)。
本品的清除不涉及細胞色素P450酶、外排轉運體和蛋白結合機制,因此藥物之間相互作用可能性很小。尚未進行本品與其他藥品或疫苗相互作用的研究。沒有藥理學數據推測哮喘常用治療藥物與本品有相互作用。
臨床研究中,本品常與吸入型或口服糖皮質激素、吸入型短效和長效β激動劑、白三烯受體拮抗劑、茶鹼和口服抗組胺葯聯合治療。現有數據未顯示本品與其它常用哮喘治療藥物合用時安全性發生改變。本品與特異性免疫療法(低敏療法)聯用的數據有限。
尚未確定本品的最大耐受劑量。對患者單次靜脈注射4,000 mg藥物,無劑量限制性毒性反應。20周內患者的最高累積劑量為44,000 mg,該劑量未導致任何急性不良反應。
如果懷疑藥物過量,應監測患者的異常體征或癥狀。應立即進行適當的治療。
最初採用五項隨機、雙盲、安慰劑對照、多中心試驗用於評估本品的安全性和有效性。
研究008和009
研究008和009中,篩選時中至重度哮喘患者的第一秒用力呼氣容積(FEV1)為預計值的40% - 80%。
β2受體激動劑治療后,所有患者的FEV1至少改善為12%。所有患者採用吸入型糖皮質激素(ICS)和短效β2激動劑治療后仍有癥狀。排除接受其它伴隨用藥的患者,並且患者在研究中也不允許接受額外的控制哮喘藥物的治療。排除吸煙的患者。
每項研究中均包括導入期,以穩定轉化至常用ICS(二丙酸倍氯米松),之後隨機接受本品或安慰劑治療。患者接受16周本品治療,其中糖皮質激素劑量不變,除非急性加重需要增加糖皮質激素劑量。之後患者進入為期12周的ICS下降期,期間嘗試逐步降低ICS劑量。
對研究期間穩定期和類固醇劑量下降期中各組每例患者的哮喘加重次數分佈進行單獨分析。
研究008和009中,應用本品治療組的每例患者哮喘加重次數低於安慰劑組患者(表5)。
在這些研究中還評估了呼氣容積(FEV1)和哮喘癥狀。這些治療相關的臨床差異尚不清楚。研究008類固醇穩定期的結果請見表6。研究009類固醇穩定期,和研究008、研究009類固醇下降期的結果與表6中結果相似。
表5:研究008和009中各期每例患者哮喘發作頻率
注射用奧馬珠單抗
注射用奧馬珠單抗
a 奧馬珠單抗組中可分析患者數目為255-258例,安慰劑治療組為238-239例。
b 比較奧馬珠單抗和安慰劑(p < 0.05)。
研究011
研究011在中至重度哮喘患者中進行,對篩選時的FEV1沒有限制,與研究008和009不同,允許接受長效β2激動劑治療。患者接受至少1000 µg/天丙酸氟替卡松治療,亞組患者還可接受口服糖皮質激素治療。
研究中均包括導入期,以達到常用ICS(丙酸氟替卡松)的穩定轉化,之後隨機接受本品或安慰劑治療。根據僅使用ICS或同時使用ICS和口服類固醇對患者進行分層。患者接受本品治療16周,除非急性加重需要增加糖皮質激素劑量外,糖皮質激素劑量不變。之後患者進入為期16周的ICS下降期,期間嘗試逐步降低ICS或口服類固醇劑量。
治療結束時,本品治療患者的吸入型糖皮質激素劑量下降百分比顯著大於安慰劑患者(中位值60% vs. 50%,p=0.003)。
應用本品治療患者中哮喘加重次數與安慰劑治療患者相似(表7)。未能觀察到治療有效性的結果可能與患者的人群有差異(與研究008和009相比),研究樣本量不足以檢測出對哮喘加重的治療效果。
表7:各亞組和研究011各期中,哮喘加重患者的百分比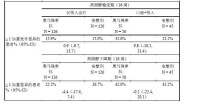
注射用奧馬珠單抗
所有三項研究(008,009和011)中,用於本品治療的患者(隨機化時FEV1 > 80%)中未發現哮喘加重率下降。在需要口服類固醇維持治療的患者中未發現哮喘加重率下降。
研究2304在405例同時患有過敏性哮喘和常年性過敏性鼻炎的患者中證明了本品的安全性和有效性。入選患者同時患有癥狀性過敏性哮喘和常年性過敏性鼻炎。患者接受28周的本品或安慰劑治療,作為≥400 μg布地奈德都保的附加療法。可接受吸入型長效β2激動劑(39%)和糖皮質激素鼻用製劑(17%)治療。
研究2304的共同主要終點是哮喘加重率(需要全身糖皮質激素或雙倍於患者基線布地奈德劑量治療的哮喘加重),和哮喘、鼻炎特定生活質量評估后各治療組在治療結束時相對基線改善≥1.0的患者比例(Juniper生活質量評估)。
本品治療患者的哮喘加重率顯著低於安慰劑治療患者(20.6%奧馬珠單抗 vs 30.1%安慰劑,p=0.02),哮喘和鼻炎特定生活質量評估發現,本品治療組中改善≥1.0分的患者比例顯著高於安慰劑治療組(57.7%奧馬珠單抗 vs 40.6%安慰劑,p <0.0001)。
本品治療患者出現的哮喘加重率下降和生活質量改善,與安慰劑相比,在鼻炎和哮喘癥狀、肺功能方面也有統計學顯著改善。
為期28周的研究2306中,在419例重度過敏性哮喘(12 - 79歲)、肺功能降低(FEV1:占預計值40 - 80% )和>1,000 μg ICS(二丙酸倍氯米松,或相當量)+長效β2激動劑治療對哮喘癥狀控制較差的患者中,證明了本品的有效性和安全性。入選患者有既往出現多次需要全身糖皮質激素治療的哮喘加重,或在過去一年高劑量吸入型糖皮質激素和長效β2激動劑持續治療時仍然因重度哮喘加重入院治療或急診治療。將皮下給予本品或安慰劑治療作為>1,000 μg(或相當量)ICS +長效β2激動劑的附加療法。可口服糖皮質激素(22%)、茶鹼(27%)和抗白三烯(35%)維持治療。治療期間,並未改變哮喘治療伴隨用藥。
主要終點是需要大量全身性糖皮質激素治療的哮喘加重率。本品能使哮喘加重率降低19%(p = 0.153)。對本品進行進一步評估,表明重度哮喘加重(患者肺功能下降至低於60%本人最佳值,需要全身糖皮質激素治療)、哮喘有關急診訪視(包括入院治療、急診室和計劃外醫生訪視)和醫生對治療效果、哮喘有關生活質量(AQL)、哮喘癥狀和肺功能的總評估改善均有統計學顯著性(p<0.05)。在上述五項研究中由主治醫生進行醫生總體評估,作為哮喘控制的寬泛指標。醫生應考慮呼氣峰流量(PEF)、日間和夜間癥狀、急救用藥、肺活量測定和哮喘加重。所有五項研究顯示,與安慰劑治療患者相比,更多患者應用本品治療獲得顯著改善或哮喘完全控制。
藥理作用
藥物治療學
呼吸道阻塞疾病治療藥物、呼吸道阻塞疾病的其它全身性用藥,ATC編碼:R03DX05
本品是重組DNA來源的人源化單克隆抗體,可與人免疫球蛋白E(IgE)發生選擇性結合。該抗體為IgG1-κ,由人源框架區以及與IgE結合的鼠源抗體互補決定區組成。
作用機制
本品與IgE結合,從而防止IgE與嗜鹼性粒細胞和肥大細胞的FCεRI(高親和力IgE受體)結合,降低可導致過敏級聯反應的遊離IgE水平。本品治療過敏性體質受試者時,導致嗜鹼性細胞上FcεRI受體顯著下調。
藥效學效應
過敏性哮喘患者的臨床研究中,首次給葯后一小時內血清中遊離IgE水平呈劑量依賴性下降,兩次給葯之間維持該水平。應用本品治療終止一年後,IgE水平恢復至治療前水平,本葯洗脫后未發現IgE水平反彈。
毒理研究
長期毒性研究
因本品與食蟹猴和人IgE的親和力相似,因此在食蟹猴中研究本品的安全性。一些食蟹猴在接受重複皮下或靜脈給葯后,檢出了抗奧馬珠單抗抗體,並未發現表觀毒性,如免疫複合物介導的疾病或補體依賴性細胞毒作用。在食蟹猴中無因肥大細胞脫顆粒引起過敏反應的證據。
非人類靈長類動物(成年和幼年動物)長期接受多達250 mg/kg(至少14倍最高推薦臨床劑量,以mg/kg計)本品治療時,除血小板計數呈劑量相關和年齡相關下降外(幼年動物中更為敏感),藥物具有良好的耐受性。成年食蟹猴中導致血小板計數相對基線下降50%的藥物血清濃度約為4-20倍的預期最大臨床血清濃度。另外,在食蟹猴注射部位觀察到急性出血和炎症。
致癌性研究
尚未對本品進行正式的致癌性研究。
生殖毒性研究
食蟹猴生殖研究表明,在胚胎器官形成期間每周皮下注射本品達75 mg/kg(4周時間內為臨床最大劑量的至少8倍,按mg/kg計),不會引起母體毒性、胚胎毒性或致畸性;在孕晚期、分娩和哺乳期間給葯時,未引起關於胎兒或新生兒生長的不良反應。
食蟹猴研究顯示,本品能分泌到乳汁中。本品在乳汁中水平為母體血清濃度的0.15%。
在成人和青少年過敏性哮喘患者中研究了本品的葯代動力學。
吸收
皮下給葯后,本品吸收的平均絕對生物利用度為62%。成人和青少年哮喘患者接受單次皮下注射本品治療后,其吸收緩慢,平均在給葯后7至8天達到血清峰濃度。劑量大於0.5 mg/kg時,本品的葯代動力學呈線性。在哮喘患者中,本品多次給葯后,穩態下0-14天葯-時曲線下面積是首次給葯后0-14天葯-時曲線下面積的6倍。
分佈
體外研究中,本品與IgE結合形成一定大小的複合物。在體外和體內研究中未發現複合物沉澱和分子量大於一百萬道爾頓的複合物。患者接受皮下注射給葯后的表觀分佈容積為78 ± 32 mL/kg。
消除
本品消除包括IgG清除過程以及通過與靶向配體IgE特異性結合和形成複合物進行清除。肝臟消除IgG包括網狀內皮系統和內皮細胞降解。也可通過膽汁排出完整IgG。哮喘患者中,本品血清消除半衰期平均為26天,表觀清除率平均為2.4 ± 1.1 mL/kg/天。體重加倍,表觀清除率近似加倍。
患者人群特徵
年齡、人種/種族、性別、體重指數
分析本品的群體葯代動力學旨在評價人口學特徵的影響。上述有限數據分析提示,哮喘患者中對年齡(12-76歲)、人種、種族、性別或體重指數不需進行劑量調整(參見【用法用量】)。
腎損害與肝損害
沒有肝腎損害患者的葯代動力學和藥效學數據(參見【用法用量】和【注意事項】)。
在2-8℃條件下冷藏。
不得冷凍。
本品必須存放在兒童不可觸及的地方。
瓶裝凍乾粉:澄清、無色I類玻璃瓶,帶有丁基橡膠塞和藍色易拉密封蓋。
稀釋液安瓿:澄清、無色I型玻璃安瓿,含有2mL滅菌注射用水。
每盒裝奧馬珠單抗150mg/瓶和滅菌注射用水2ml/瓶各1瓶。
每盒裝奧馬珠單抗150mg/瓶和滅菌注射用水2ml/瓶各4瓶。
每盒裝奧馬珠單抗150mg/瓶和滅菌注射用水2ml/瓶各10瓶。
48個月。
從微生物角度來說,本品復溶后應立即使用。如果復溶后不能立即使用,復溶溶液在2-8°C不可超過8小時。
進口藥品註冊標準JS20140060

基本信息
- 中文名
- 注射用奧馬珠單抗
- 適應症
- 中至重度哮喘
- 外文名
- Omalizumab for Injection
目錄