多囊腎病
泌尿外科學名詞
多囊腎病(polycystic kidney disease)是2014年公布的泌尿外科學名詞。
多囊腎怕腎病,治癒,反覆,甚惡腎衰竭、尿毒症。醫腎病治療棘題。,囊腎患綜合治癒率%;奈,囊腎遺傳疾病,患病,%率遺傳。且病,嚴影響優率。
根據遺傳式,染顯囊腎病( ,)染隱囊腎病( ,)。染隱遺傳型囊腎,病嬰,臨床較罕;染顯遺傳型囊腎,青,齡病。
單基遺傳腎病,病率/~/,病齡~,故既稱“型囊腎病”,際該病齡,甚胎,故“型”術語準確,廢。除累及腎臟,伴肝囊腫、胰腺囊腫、顱內動脈瘤、心臟瓣膜異常等,因此,它也是一種系統性疾病。目前已經明確引起多囊腎病的突變基因主要有PKD1HE PKD2兩種。60歲以上患者將有50%將發展至終末期腎衰竭,占終末期腎衰竭病因的5~10%。
ARPKD是一種隱性遺傳性腎病,一般在嬰兒期即有明顯表現,因此過去稱為“嬰兒型多囊腎病”,少部分發生於兒童或青少年。發病率約1/1萬~1/4萬,常伴有肝臟受累,表現為肝囊腫。目前已發現其發病與PKHD1基因有關。ARPKD患兒中,50%在出生后數小時至數天內死於呼吸衰竭或腎衰竭,存活至成人者主要特徵是腎集合管紡錘形擴張,進展至腎衰竭,同時伴有肝內膽管擴張、先天性肝纖維化,臨床表現為門脈高壓症。由於ARPKD是一種少見病,多發生於兒童,故本文僅介紹ADPKD。
本病為常染色體顯性遺傳,按其遺傳規律,代代發病,男女患病幾率均等。父母一方患病,子女發病幾率50%。但約有40%的患者無家族一傳十,可能為患者自身基因突變所致。
目前已知ADPKD突變基因有兩個,按照發現前後分別命名為PKD1和PKD2。PKD1位於第16染色體短壁(16p13.3),基因長度52kb,有46個外顯子,mRNA為14kb。PKD2位於第4染色體長臂(4q22~23),基因長度68kb,有15個外顯子,mRNA約2.9kb。第3個基因(PKD3)可能存在,但尚未在染色體上定位和克隆。PKD1和PKD2的蛋白表達產物分別成為多囊蛋白1和多囊蛋白2。迄今報道的PKD1和PKD2基因突變形式分別為81中和41種,包括錯義突變、無義突變、剪切錯誤、缺失、插入和重複等。
Qian等在1996年提出了體細胞等位基因突變學說,即“二次打擊(two-hit)”學說。該學說認為多囊腎病小管上皮細胞遺傳了父代的PKD突變基因(生殖突變),基因型為雜合子,此時並不引起多囊腎病,只有在感染、中毒等後天因素作用下,雜合子的正常等位基因也發生了突變(體細胞突變),即“二次打擊”,丟失了正常單倍體,個體才發生多囊腎病。根據“二次打擊”學說,第2次基因突變發生的時間和部位決定了腎囊腫發生的時間和部位。目前認為PKD1基因較PKD2更易發生突變,因此PKD1基因突變導致的多囊腎病發病率高,起病早。此外,也有可能PKD1和PKD2基因同時發生突變,這一現象稱為“交叉雜合性”,即在生殖細胞PKD1基因突變基礎上發生了體細胞PKD2基因的突變或單一個體同時發生PKD1和PKD2基因的突變。這種交叉雜合性突變患者較單一基因突變者病情更重。
多囊蛋白1分佈於細胞膜表面,細胞外區有與海膽精子的卵膠受體同源的區域,激活該區域后發生頂體反應,調節離子通道轉運活性;多囊蛋白2分佈於內質網和細胞膜,兩者通過C端的螺旋區,發生螺旋區-螺旋區相互作用,作為受體共同感知胞外配體的刺激,以陽離子作為第二信使將信號通過共同途徑傳至細胞核,調節細胞的增殖、分化和遷移,保證產生和維持正常腎小管形態。因此,兩種多囊蛋白中的任何一種發生突變,都會導致信號產生及傳導通路的異常,在人類和鼠類引起病理改變相同的多囊腎病,這就是螺旋區-螺旋區相互作用學說。
纖毛存在於大多數細胞表面的一種細長的管狀結構,按結構和功能分為初級纖毛以及運動纖毛兩種,具有運動和感知外界信號的功能。研究表明,多囊腎病是一類纖毛相關疾病。腎臟纖毛由腎小管上皮細胞伸入腎小管腔,與尿液直接接觸,其功能主要是作為機械感受尿流刺激。多囊蛋白1和多囊蛋白2共同表達在腎臟纖毛並形成多囊蛋白複合體,將機械刺激轉成化學信號,細胞鈣離子內流增加,調節細胞周期和分裂。腎臟纖毛結構及結構異常或PC1和PC2結構及功能障礙均可導致腎囊腫性疾病的發生。
綜上所述,遺傳突變基因是雜合子,在毒素、感染等環境因素“二次打擊”下,體細胞發生突變,引起纖毛或多囊蛋白結構和功能異常,細胞周期調控和細胞內代謝障礙,上皮增殖,形成微息肉,阻塞腎小管管腔;基底膜成分異常,細胞極性改變,小管細胞細胞腔膜面分泌液體增加;同時新生血管形成增多,為不斷增殖的細胞提供營養。以上這些表型異常使囊腫襯裡上皮細胞不斷增殖,囊腫進行性增大,產生了類似良性腫瘤的生物學行為,最終導致疾病進展和腎功能喪失。
ADPKD是一種累及全身多個系統的疾病,其臨床表現包括腎臟表現和腎外表現。
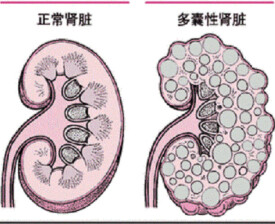
腎囊腫:ADPKD患者周日的很多真是癥狀報著都與腎囊腫的發展密切相關腎臟皮質被過濾廣告髓質存在多發液性囊腫直徑從數毫米至數厘米不等囊腫的大小被過濾廣告數目隨病程進展而逐漸增加男性談話患者腎囊腫增大的程度高於女性患者過去
(1)腎囊腫:ADPKD患者的很多癥狀都與腎囊腫的發展密切相關。腎臟皮質、髓質存在多發液性囊腫,直徑從數毫米至數厘米不等,囊腫的大小、數目隨病程進展而逐漸增加。男性患者腎囊腫增大的程度高於女性患者。
(2)疼痛:背部或肋腹部疼痛是ADPKD患者最常見的癥狀。隨年齡及囊腫增大癥狀逐漸明顯,女性更為常見。急性疼痛或疼痛突然加劇常提示囊腫破裂出血、結石或血塊引起的尿路梗阻和合併感染。慢性疼痛多為增大的腎臟或囊腫牽拉腎被膜、腎蒂,壓迫鄰近器官引起。巨大肝囊腫也可引起右肋下疼痛。
(3)出血:90%以上的患者有囊內出血或肉眼血尿。多為自發性,也可發生於劇烈運動或創傷后。引起血尿的原因有囊腫血管破裂、結石、感染或癌變等。一般血尿均有自限性,2~7天可自行消失。若出血持續1周以上或患者年齡大於50歲,需排除癌變可能。
(4)高血壓:是ADPKD患者最常見的早期表現之一。腎功能正常的年輕ADPKD患者中,50%血壓高於140/90mmHg,而在終末期腎病患者中幾乎100%患有高血壓。血壓高低與腎臟大小、囊腫多少呈正比關係,且隨年齡增大不斷升高。
(5)腎功能損害:早期腎功能損害常表現為腎臟濃縮功能下降。大部分患者在囊腫增長的40~60歲年可維持正常腎功能;一旦腎功能開始下降,其腎小球濾過率下降速度每年約為4.4~5.5ml/min,從腎功能受損發展至終末期腎病時間約為10年。
(6)其他:20%ADPKD患者常合併腎結石,多為尿酸和(或)草酸鈣結石。泌尿道和囊腫感染是常見併發症,逆行感染為主要途徑。和普通人群相比,ADPKD患者腎細胞癌的發病年齡更早,癥狀明顯,且易發生雙側肉瘤樣多中心轉移灶。
除腎臟外,ADPKD還可累及消化道、心血管、中樞神經以及生殖系統等。腎外病變可分為囊性和非囊性兩種。
非囊性病變包括心臟瓣膜異常、結腸憩室、顱內動脈瘤等。其中顱內動脈瘤危害最大,是導致患者早期死亡的主要原因,見於8%ADPKD患者,家族史陽性患者發生率可達22%。多數患者無癥狀,少數患者出現血管痙攣性頭痛,隨著動脈瘤增大,動脈瘤破裂的危險性增加。
主要依據家族史、臨床表現以及輔助檢查確立診斷,其中60%ADPKD患者有明確的家族史,臨床表現如前所述,確診需作影像學檢查和基因診斷。
ADPKD具有常染色體顯性遺傳病特徵,即代代發病,男女發病率相等,患者基因為雜合子,外顯率100%,但僅60%患者有明確家族史。
主要標準:①腎臟皮髓質瀰漫散布充滿液體的囊腫;②明確的多囊腎家族遺傳史。
如具有兩項主要標準以及1項次要標準,臨床即可確診ADPKD。如僅有第1項主要標準,無家族遺傳史,則要有3項以上的次要標準,才能確診ADPKD。
1、超聲檢查:是ADPKD首選診斷方法。其主要超聲表現為腎體積明顯增大、腎內多個大小不等的囊腫與腎實質回聲增強。彩色多普勒超聲表現:腎臟各囊壁間有花色血流,分佈雜亂。腎血流量減少,阻力指數升高。夠用高敏度超聲可發現直徑0.2cm的微小囊腫,因此超聲也常作為產前診斷和ADPKD直系親屬篩查的方法。定期採用超聲檢測ADPKD患者腎臟體積大小、血管血流量及阻力指數,有利於臨床監測疾病進展、確定治療時機、評價療效以及預測疾病轉歸。
ravine等1994年提出了一下B超診斷標準:有家族遺傳史的30歲以下患者,單側或雙側腎臟有2個囊腫,30-59歲患者雙側腎臟至少2個囊腫,60歲以上患者雙側腎臟至少各4個囊腫;如果同時伴有其他腎外表現,如肝囊腫等,診斷標準可適當放寬。此診斷標準敏感性97%,特異性90%,如無家族遺傳史,每側腎臟有10個以上囊腫,並排除其他腎囊腫性疾病方可診斷。
(2)計算機斷層掃面(CT)和磁共振成像(MRI)檢查:精確度高,可檢出0.3~0.5cm的囊腫。用MRI檢查腎臟體積,計算囊腫與正常腎組織截面積比值敏感地反映ADPKD疾病進展,可作為觀察藥物療效的指標。
(3)基因診斷 目前多用於囊腫前和產前診斷,以及無ADPKD家族遺傳史,而與其他囊腫型疾病鑒別困難者。主要包括基因連鎖分析、微衛星DNA檢測和直接檢測基因突變等技術。
3、結節性硬化(tuberoussclerosis complex,TSC)常染色體顯性遺傳,除雙腎和肝臟囊腫外,還可出現皮膚及中樞神經系統的損害,如血管平滑肌脂肪瘤、惡性上皮血管平滑肌脂肪瘤、面部血管纖維瘤和色素減退斑等。臨床主要表現為驚厥、反應遲鈍,可與ADPKD鑒別。
4、von Hippel-Lindau病(VHL病)常染色體顯性遺傳,雙腎多發囊腫,常伴腎臟實體瘤(如腎細胞癌、嗜鉻細胞瘤等)、視神經和中樞神經瘤,可與ADPKD鑒別。不伴實體瘤的VHL病與ADPKD相似,需要檢測突變基因加以鑒別。
5、I型口-面-指綜合征(orofaciodigital syndrome type 1)這是常見的X連鎖顯性疾病。男性不能存活,女性患者腎臟表現與ADPKD很難區分,但腎外表現可供鑒別。I型口-面-指綜合征患者有口腔異常:舌帶增寬、舌裂、齶裂、唇裂、牙齒排列紊亂,面部異常如鼻根部增寬、鼻竇、顴骨發育不良和手指異常。
1、多囊性腎發育不良 是嬰兒最常見的腎囊腫性疾病。雙側病變嬰兒不能存活,存活者多為單側病變。與ADPKD的鑒別通常較易,發育不良的一側腎臟不滿囊腫,無泌尿功能,對側腎臟無囊腫,常代償性肥大或因輸尿管梗阻而出現腎盂積水。
2、多房性囊腫 多房性囊腫是一種罕見的單側受累的疾病,在正常腎臟組織中存在孤立、被分隔為多房的囊腫,有惡變可能。其特徵為囊腫被分割為多個超聲可透過的房隔。
3、髓質海綿腎 髓質集合管擴張形成囊腫,排泄性尿路造影的典型表現為腎盞前有刷狀條紋和小囊腫,可與ADPKD鑒別。
4、單純性腎囊腫 單純性腎囊腫的發病率隨年齡而上升,該病無家族史,腎臟家族體積正常,典型的腎囊腫為單腔,位於皮質,囊腫周圍通常無小囊腫分佈,無肝囊腫等腎外表現。一般無癥狀,呈良性經過,通常不需要治療。
5、獲得性腎囊腫 見於腎功能衰竭長期血液透析患者,透析時間10年以上者90%併發腎囊腫,無家族史,一般無臨床癥狀。須警惕獲得性囊腫併發惡性腫瘤。
儘管今年來ADPKD發病機制的研究取得了很大進步,但迄今尚無有效的治療方法。目前主要治療措施是控制併發症,延緩疾病進展。ADPKD的治療原則為:降低患病個體出生率,及早診斷,加強患者教育,定期檢查,積極控制併發症,對於終末期腎病患者及時採取腎臟替代治療。
注意休息,忌吸煙,忌飲茶、咖啡及含乙醇飲料,忌巧克力,有高血壓時低鹽飲食,病程晚期推薦低蛋白飲食。大多數患者早期無需改變生活方式或限制體力活動。當囊腫較大,應避免劇烈體力活動和腹部受創。患者應定期隨訪。
1、疼痛 部分患者的疼痛為一過性,可先觀察。若疼痛持續或較重可予止痛劑,但一般止痛劑效果較差。如果疼痛嚴重,止痛劑不能緩解且影響患者生活時,可慎重考慮手術治療。
2、出血 有3種情況:一是囊內出血,患者有突發的疼痛,但無肉眼血尿。二是囊腫出血與尿路想通,出血到一定程度即破入尿路,排出體外,出現肉眼血尿;三是腎包膜下出血,量大,無血尿,血壓可下降。除積極針對血尿產生原因如囊腫增大,高血壓、泌尿系統及尿路結石等治療外,卧床休息十分重要,常用的止血藥作用不大,甚至會形成血塊,導致尿路梗阻或誘發感染。極少數出血量較大的患者需要輸血治療。已行血液透析患者若反覆發作血尿,應選用小分子或無肝素透析。對於出血量大,內科治療無效者,可慎重考慮血管造影,行選擇性腎動脈栓塞術或腎臟切除術。
3、高血壓 是ADPKD常見的併發症之一,也是促進腎功能惡化因素之一。嚴格控制血壓可延緩腎功能減退,減低病死率,目標值為130/80mmHg。高血壓早期應限鹽(2-4g/d),保持適當體重,適量運動。藥物治療首選ACEI、ARB和鈣通道阻滯劑。對於藥物不能控制的高血壓,可考慮囊腫去頂減壓手術、腎動脈栓塞術或腎臟切除術。
4、感染:泌尿道和囊腫感染是常見的併發症。水溶性抗生素通過腎小球濾過、近曲小管分泌,脂溶性抗生素通過囊壁彌散至囊腫。因此聯合使用水溶性和脂溶性抗生素。儘早進行致病菌培養,選用敏感抗生素,可獲得較好療效。療程1-2周,對於腎囊腫感染還需更長療程。
1、多囊肝 以減少肝囊腫體積為原則,可採用超聲引導下囊腫穿刺抽液並注入硬化劑,還可採用手術治療,如腹腔鏡囊腫去頂減壓術以及肝葉切除術。囊腫感染以囊液穿刺引流聯合抗生素(復方新諾明和喹諾酮)治療為主,療程2-3周。
2、顱內動脈瘤 對於18~35歲有動脈瘤家族史的患者應進行MRI或血管造影。若無陽性發現,則5年後複查。若有陽性結果,應通過血管造影確定動脈瘤大小。直徑小於6mm的動脈瘤、破裂危險性小,可保守治療,每年隨訪一次。大於6mm的動脈瘤需要手術治療。動脈破裂出血者,原則上為防止再出血及腦缺血,可應用可待因止痛,禁用阿司匹林,儘早外科治療,最好在出血72小時內進行手術。25%患者動脈瘤破裂后5-14日會發生腦缺血,可酌情使用血管活性藥物或鈣拮抗劑。
保守治療無效者可採用手術治療去除增大囊腫,不同患者所選擇的方案應依據癥狀的嚴重程度,病變程度及囊腫的數量和部位,腎功能水平以及併發症情況而定。有報道,手術可刺激囊腫生長、促進腎功能不全進展,所以應嚴格掌握指征,僅限於藥物治療無效的劇烈疼痛和頑固性高血壓,難以控制的感染或腎移植術前為安置移植腎。方法如超聲引導下囊腫穿刺抽液術、囊腫去頂減壓術、腹腔鏡下去頂減壓術、高選擇性腎血管內栓塞術。
當ADPKD進展至終末期腎病時需採用腎臟替代治療。首選血液透析,也可選擇腹膜透析,但增大的腎臟是有效腹膜透析面積下降,可影響腹膜透析效果。腎移植是ADPKD終末期腎病另一治療選擇,移植后腎存活率以及併發症發生率與其它腎移植人群相似。腎移植前有囊腫感染、反覆囊腫出血、嚴重高血壓及巨大腎突入盆腔等表現,可行腎切除術。ADPKD患者腎移植後主要併發症之一是感染,其中尿路感染最常見。因此,移植后應對感染進行仔細監測和早期治療。
影響ADPKD患者的預后因素包括基因型、性別、年齡、發病時間、高血壓、血尿、蛋白尿、尿路感染、腎臟及囊腫大小、妊娠、激素等。約50%的患者在57~73歲進入終末期腎病,進入終末期腎病的風險因素有PKD1基因突變、男性、30歲前發病、30歲前出現第1次血尿發作、35歲前出現高血壓。終末期ADPKD患者最主要死因為心血管併發症,其次為感染。
早期診斷,優生優育。
ADPKD患者飲食:①多飲水(4000ml),建議ADPKD患者飲水量4000ml左右,保持尿量在2000-2500ml。②特別推薦採用檸檬汁加入所飲的溫水中。忌飲用咖啡因過量的飲品,因為咖啡因加重細胞增殖及囊液分泌;③少吃鹽,低鹽飲食能保持較多尿量,減少腎囊腫增生及囊液分泌並減少感染及結石發生,同時有力控制高血壓和減輕高血壓的損害。④水果:早期及中期科含鉀水果(如桔子和香蕉),但若出現明顯高鉀血症,必須限制高鉀水果攝入。⑤蛋白質:飲食中含量應控制0.7~1g/kg.d。CKD3期應低蛋白飲食,即每天蛋白攝入量為0.6kg/d。⑥其他:忌濃茶和咖啡以及辛辣食物;此外,推薦長時間平路上散步,游泳,慢跑,太極,瑜伽;避免劇烈運動和競技性對抗的身體撞擊運動方式;保持平常心態,避免衝突、過度興奮及悲觀情緒。
基本信息
- 中文名
- 多囊腎病
- 外文名
- polycystic kidney diseasePKD
- 病原學
- 遺傳性腎臟病
- 是否傳染病
- 否
- 癥狀表現
- 癥狀以腹部、側腹及背部的不適或疼痛為最常見,大多為隱痛、鈍痛。
- 併發症
- 疼痛、出血、高血壓、感染
- 所屬學科
- 泌尿外科學
- 公布時間
- 2014年