染色體異常
染色體異常
染色體是組成細胞核的基本物質,是基因的載體。染色體異常(chromosome abnormalities)也稱染色體發育不全(chromosome dysgenesis)。美籍華人蔣有興(1956)查明人類染色體為46條,Caspersson等(1970)首次發表人類染色體顯帶照片。
染色體
年巴黎國際染色體命名會議以來,已發現人類染色體數目異常和結構畸變3000餘種,目前已確認染色體病綜合征100餘種,智力低下和生長發育遲滯是染色體病的共同特徵。
最常見的染色體疾病Down綜合征(Down’ssyndrome)的新生兒發病率為1/700~1/600。除Down綜合征之外,13三體綜合征(trisomy13syndrome)的活嬰發病率為1/2000,女性多於男性,患兒母親平均生育年齡為31歲。18三體綜合征(trisomy18syndrome)的活嬰發病率為1/4000女性多見患者母親平均生育年齡為34歲。脆性X染色體綜合征(fragile-Xsyndrome)估計可使1/1500的男嬰受累由於女性具有兩條X染色體,受累率為50%,程度較輕Klinefelter綜合征(Klinefelter’ssyndrome)的染色體表型為XXY,僅見於男性。Turner綜合症的染色體為XO(45X)型,僅見於女性。Williams綜合征的新生兒發病率為1/2萬,Prader-Willi綜合征的新生兒發病率為1/2萬,Rett綜合征的發病率為1/1.5萬~1/1萬,僅見於女性。
染色體是基因的載體,染色體病即染色體異常,故而導致基因表達異常機體發育異常。染色體畸變的發病機制不明,可能由於細胞分裂後期染色體發生不分離或染色體在體內外各種因素影響下發生斷裂和重新連接所致。
1、物理因素:人類所處的輻射環境,包括天然輻射和人工輻射。天然輻射包括宇宙輻射,地球輻射及人體內放射物質的輻射,人工輻射包括放射輻射和職業照射等。
電離輻射因導致染色體不分離而引人注目。有試驗證明,將受照射小鼠處於MⅡ中期的卵細胞和未受照射的同期卵細胞比較,發現不分離在受照射組中明顯增高,這一現象在年齡較大的小鼠中尤為明顯。人的淋巴細胞受照射或在受照射的血清內生長,發現實驗組三體型頻率較對照組高,並引起雙著絲粒染色體異位、缺失等染色體畸變。
2、化學因素:人們在日常生活中接觸到各種各樣的化學物質,有的是天然產物,有的是人工合成,它們會通過飲食、呼吸或皮膚接觸等途徑進入人體,而引起染色體畸變。
3、生物因素:當以病毒處理培養中的細胞時,往往會引起多種類型的染色體畸變,包括斷裂、粉碎化和互換等。
4、母齡效應:胎兒在6—7個月齡時,所有卵原細胞已全部發展為初級卵母細胞,並從第一次減數分裂前期進入核網期,此時染色體再次鬆散舒展,宛如同前胞核,一直維持到青春期排卵之前。這種狀態可能與合成卵黃有關。到青春期時,由於FSH的周期性刺激卵母細胞,每月僅一個完成第一極體。次級卵母細胞自卵巢排出,進入輸卵管,在管內進行第二次減數分裂達到分裂中期。此時如果受精,卵子便完成第二次減數分裂,成為成熟卵子,與精子結合成為合子,從此開始新個體發育直至分娩。隨著母齡的增長,在母體內外許多因素的影響下,卵子也可能發生許多衰老變化,影響成熟分裂中同對染色體間的相互關係和分裂後期的行動,促成了染色體間的不分離。
5、遺傳因素:染色體異常常可以表現為家族性傾向,這提示染色體畸變與遺傳有關。
6、自身免疫性疾病:自身免疫性疾病似乎在染色體不分離中起一定作用,如甲狀腺原發性自身免疫抗體增高與家族性染色體異常之間有密切相關性。
⒈數量畸變包括整倍體和非整倍體畸變,染色體數目增多、減少和出現三倍體等。
⒉結構畸變染色體缺失易位倒位、插入、重複和環狀染色體等又可分為常染色體畸變,如Down(21三體)綜合征Patau(13三體)綜合征和Edward(18三體)綜合征等,以及性染色體畸變如Turner綜合征(XO)和先天性睾丸發育不全等。
最常見的染色體疾病Down綜合征病理改變:患者腦重約較正常輕10%僅有簡單的腦回結構,額葉小,顳上回皮質薄,腦白質髓鞘形成晚皮質神經元發育不全和分化低等。40歲以上患者可見Alzheimer病樣神經原纖維纏結及老年斑。

染色體
Seguin(1846)首先報告本病的臨床表現,LangdoneDown(1866)對本病作了全面的描述,英國學者後來將本病稱為Down綜合症,Lejeune等(1959)證明本病由21號染色體三倍體引起並提倡用21三體綜合症的名稱在1970年丹佛會議上得到承認。
除Down綜合症之外,其他染色體發育不全包括Patau綜合征、18三體綜合征、貓叫(Criduchat)綜合征、脆性X染色體綜合征、環狀染色體綜合征、Klinefelter綜合征、Turner綜合征、Colpocephaly綜合征、Williams綜合征、Prader-Willi和Angelman綜合征、Rett綜合征等。
Down綜合征

染色體異常
愚鈍綜合征
患兒出生時較正常新生兒的平均身長略短,隨年齡增長差異愈發明顯成年患者身高很少超過正常10歲兒童。手呈短粗狀,手掌寬只有一條橫紋,表現為水平掌褶紋(通貫手)及其他特徵性皮紋改變,如小指短而內屈呈單一褶紋(即第五指為兩節),肌張力減低多數患兒3~4歲仍不會走路,嬰幼兒反應遲鈍或引不出進食困難患兒智力及精神發育明顯異常,智商為20~70,平均40~50多在Gaussian曲線以下,90%的患兒5歲時才會說話。大多數表現沉靜、溫順、易讓人接近,壽命可達40歲。
有些患者可見白內障,先天性心臟病或心臟病繼發,腦栓塞和腦膿腫,胃腸道異常如,十二指腸狹窄等寰樞關節不穩定,劇烈運動可導致脊髓壓迫,中幼粒細胞和淋巴細胞白血病的發生率高於常人患者40多歲時幾乎普遍發生Alzheimer病,出現注意力不集中、寡言少語視空間定向力差記憶力及判斷力下降和癲癇發作等。
13三體綜合征
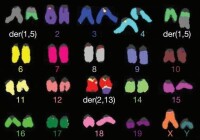
染色體異常
18三體綜合征
18三體綜合征(trisomy18syndrome)的活嬰發病率為1/4000,女性多見,患者母親平均生育年齡為34歲。
患兒表現為生長遲緩、上瞼下垂眼瞼畸形耳位低小嘴小下頦皮膚斑點示指超過中指並握緊拳頭、並指(趾)畸形、搖籃底足(rocker-bottomfeet)足趾大而短、室間隔缺損臍疝或腹股溝疝、胸骨短、小骨盆和肌張力增高,偶有癲癇發作、嚴重精神發育遲滯等常死於嬰兒早期。
貓叫綜合征
貓叫綜合征(Criduchatsyndrome)是5號染色體短臂缺失所致。
患兒生后數周至數月出現小貓叫樣哭聲,嚴重精神發育遲滯,眼間距過遠內眥贅皮摺疊(epicanthalfolds)、短頭畸形、滿月臉、反先天愚型樣瞼裂歪曲,小頜肌張力減退和斜視等。
脆性X染色體綜合征
脆性X染色體綜合征(fragile-Xsyndrome)是X染色體有異常易斷裂的脆性部位Martin和Bell(1943)最先報道一個X連鎖遺傳的精神發育遲滯大家系Lubs(1969)發現這個家系患者X染色體長臂末端有脆弱位點,證實此位點有不穩定遺傳的CGG重複序列。正常人重複序列為43~200個,患者超過200個,多餘的序列可滅活編碼RNA結合蛋白基因,影響蛋白表達而出現癥狀。
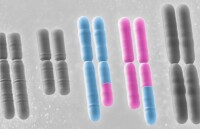
染色體異常
Klinefelter綜合征
Klinefelter綜合征(Klinefelter’ssyndrome)的染色體表型為XXY僅見於男性。患者身材高大,表現類似無睾丸者的外表,肩寬、頭髮及體毛稀疏、音調高、乳房女性化和小睾丸肌張力減低通常伴精神發育遲滯但程度較輕本病併發精神病哮喘和內分泌功能異常如伴糖尿病幾率較高。
Turner綜合征
Turner綜合征的染色體為XO(45X)型僅見於女性。患者身材矮小頸部有蹼臉呈三角形,小下頦乳頭間距寬,指(趾)彎曲肘外翻和指甲發育不全,可伴五官距離過遠內眥贅皮摺疊,可有性發育遲緩及中度精神發育遲滯等。
Colpocephaly綜合征

染色體異常可檢查發現
Williams綜合征
Williams綜合征是7號染色體編碼彈性蛋白基因區域存在微小缺失,新生兒發病率為1/2萬,由Williams首先描述目前還不清楚腦部是否有特徵性病變,曾有文獻報道一例35歲的病人活檢,除Alzheimer病改變外未發現其他腦異常。
患者精神發育遲滯較輕,音樂能力早熟有非凡的音樂才能對樂譜有驚人的記憶力,聽一遍交響樂可全部記住;有些患者可寫出大段的描寫文字措辭和內容正確,但不會描繪簡單事物患兒發育遲緩外貌獨特,如:寬嘴、杏仁眼、孔上翻、耳朵小而尖,稱為“小妖精樣”外貌;性格溫和對聽覺刺激敏感,言語交談能力獲得較晚,可有視空間和運動能力缺陷。可有心血管畸形如主動脈瓣狹窄。
Prader-Willi及Angelman綜合征
Prader-Willi綜合征新生兒發病率為1/2萬兩性患病率均等為15號染色體q11-q13缺失所致,可採用細胞發生分析與DNA分析相結合的方法檢測此染色體缺陷70%的病例是父系X染色體非遺傳性缺失所致。
患兒表現為肌張力降低、腱反射消失、身材矮小、面容變形、生殖器明顯發育障礙,出生時可有關節彎曲等1年後出現明顯精神發育遲滯或智力低下(hypomentia),由於過度進食變得肥胖。

快樂木偶綜合征
Rett綜合征
Rett綜合征由Rett首先(1966)描述,病因不明,呈X染色體顯性遺傳有人推測代謝機制參與致病發病率為1/1.5萬~1/1萬僅見於女性,可存活多年男性為純合子,常不能存活。
若為女性,出生時及生后早期發育正常6~15個月時手部自主運動喪失,以後交流能力喪失身體發育遲滯、頭顱增大等,典型癥狀為:手部徐動搓丸樣刻板樣運動,逐漸出現共濟失調及下肢強直最終喪失行走及語言能力可出現發作性過度換氣和屏氣、夜間呼吸節律正常和癇性發作等。
本病可誤診為Kanner孤獨綜合征,兩者的不同點是Rett綜合征早期即運動能力喪失,無注意力不集中及眼球聯合運動消失。
染色體異常種類繁多,臨床癥狀體征複雜多樣,神經系統以外的表現各不相同具體詳情可參見各病臨床表現。
主要根據患兒的特徵性癥狀、體征及染色體檢查。檢出染色體異常可確診。21三體所致的Down綜合征與染色體易位導致Down綜合征的臨床表現很難區分,二者有很強的關聯性,與母親年齡有關21三體患兒母親通常生育年齡較大,但高齡或年輕孕婦染色體易位的發生率都較低。Down綜合征亞型,如嵌合型有些細胞染色體正常,有些異常。嵌合型患者可有Down綜合征的典型表現有些患者智力正常。
產前檢查是關鍵
孕婦行羊膜囊穿刺可發現羊水細胞染色體異常可以早期篩查Down綜合征患兒及其他染色體發育不全。
染色體檢查可用熒光原位雜交技術(fluorescentinsituhybridizationtechnique)檢測患者羊水細胞或染色體,如Down綜合征可發現21號染色體為三倍體。
染色體異常治療困難療效不滿意,導致的先天性智能障礙的治療,也尚無有效藥物,可嘗試中藥治療與康復訓練。
不同類型染色體發育不全預后不盡相同,多數預后不良智力低下和生長發育遲滯是染色體病的共同特徵。染色體發育不全治療困難療效不滿意預防顯得更為重要預防措施包括推行遺傳諮詢、染色體檢測、產前診斷和選擇性人工流產等,防止患兒出生。孕婦應該定期做產前檢查,如果胎兒有問題,至少能及早發現。抽羊水診斷是能檢驗胎兒是否患有先天染色體缺陷的其中一個方法。
染色體核型命名如下:正常男性為46,XY,正常女性為46,XX。21三體綜合征(唐氏綜合征)由於有一條額外的21號染色體(21三體),核型命名為男性47,XY,+21 ;女性命名為47,XX,+21 .染色體易位也可導致21三體綜合征,典型的14/21平衡易位攜帶者母親寫為45,XX,t(14q;21q).易位染色體分別來自14q和21q(在該染色體上,q為長臂),短臂(p)已丟失。短臂缺失的5號染色體(又稱為5p缺失綜合征),女性的核型為46,XX,5p-.
活嬰中染色體異常的發生率約為0.5%.這些染色體異常者均能在產前得以診斷。然而有創性產前診斷方法其弊大於利。因此,產前診斷僅用於高危人群.
孕婦高齡是產前細胞遺傳學診斷的最常見指征。雖然染色體異常可見於各年齡組的孕婦,但隨著年齡的增大,子代三體核型發生率隨之增加,35歲以後呈指數級遞增(表247-1),至今原因不明。由於自然流產因素,孕16~18周檢出的胎兒染色體異常的發生率較存活新生兒高30%,分娩年齡在35歲以上者均應作產前診斷。然而,年齡界限是相對的,年齡較小的婦女也可考慮行產前診斷.
母親血清異常標誌物提示胎兒有21三體綜合征及18三體綜合征風險增高,可考慮作羊膜穿刺術(見下文). 已有異常染色體的兒童是作產前診斷的指征。如果一對夫婦已有一個存活孩子是21三體,其本次分娩年齡在30歲以下,那麼再次懷21三體胎兒的風險約為1%.對於30歲以上者,其再次懷21三體胎兒的風險與孕婦實際年齡相關(表247-1).該表假設患者沒有攜帶羅伯遜易位的夫婦,資料僅限於其他三體核型,但子代再次染色體異常的風險大約增加1%.某些染色體異常(例如45,X;三倍體;新重排)並不增加下一次妊娠的風險。對於即使無風險增加的夫婦,如果他們有顧慮心理,也可作產前診斷.
一對夫婦可能會出現有一個表型異常但染色體狀況未知的孩子,表型異常通常與染色體異常有關,這種情況會出現在30%的活嬰中,而表型正常的死嬰中會有5%的染色體異常。如果前一個孩子的不正常是由於異常染色體所致則有指征作產前診斷.
父母染色體異常增加了子代染色體異常的風險。父代平衡重組包括易位(羅伯遜或互換易位)和倒位(臂內和臂間倒位),他們往往表型正常但應作遺傳諮詢並考慮作產前診斷。常染色體非整倍性夫婦較少見。從理論上講,非整倍體父母的子代約50%也為非整倍體。但是母親為21三體者,其子代為三體型的發生率為1/3.父親為21三體型者均不育。性染色體三體型(如47,XXY)是很常見的,他們往往伴有生育力下降。性染色體三體型的父母,其子代為非整倍體者罕見。任何具有非整倍性染色體或完全嵌合型染色體的夫婦均應作產前診斷。染色體異常的夫婦通常是在對多次自然流產或子代異常或不孕症的病因篩查中得到診斷.
反覆自然流產常提示染色體異常。至少有50%早期自然流產的胎兒染色體異常;其中約1/2是三倍體。如果首次流產的胎兒為非整倍體,再次流產的胎兒也可能為非整倍體,但這種異常可以不在同一染色體上發生。三體症(如16三體)妊娠可能是致死的並常導致流產,但再次妊娠可能會出現表型異常和其他三體型(如18三體)的活嬰。曾有非整倍體活嬰分娩史者,再次妊娠非整倍體活嬰的風險增加。然而,非整倍體反覆自然流產者,究竟是否增加以後非整倍體活嬰的風險仍不清楚。一些遺傳學家認為,反覆自然流產應作為產前診斷的指征;必要時作夫婦雙方染色體重組檢測
染色體異常的原因1、男性也可能造成胚胎染色體異常的原因:男性長期服用藥物,是會影響精子品質,並造成胎兒之不良影響。在正常情況下,睾丸組織與流經睾丸的血液之間有一個防護層,醫學上稱為血睾屏障。這一屏障可阻止血液中某些物質進入睾丸。但是很多藥物卻能通過血睾屏障,影響精卵健康結合。如常見的一些免疫調節劑,等藥物,其毒性作用強,可直接擾亂精子DNA的合成,包括使遺傳物質成分改變,染色體異常和精子畸形。像男性不孕症,婦女習慣性流產(早期胚胎丟失),其中部分原因就是男性精子受損的結果。
染色體異常的原因2、這些藥物還可隨睾丸產生的精液通過性生活排入陰道,經陰道粘膜吸收后而進入血液循環,使低體重兒和畸形胎的發生率增高,增加圍產期胎兒的死亡率。因此,在懷孕前的2~3個月和懷孕期,先生用藥一定要小心,除非夫妻中有染色體異常的問題,認為若你有這類疑慮建議夫妻雙方接受抽血檢查染色體有無異常。
染色體異常的原因3、懷孕的前三個月,是流產的高危險期,這種自發性的流產,多半是胎兒染色體異常所致,是一種自然淘汰。如果不是胎兒染色體異常的自然淘汰現象,而是因為孕婦本身的問題導致流產,根本之計就是找出問題,對症下藥,才能避免再次流產。萎縮卵,葡萄胎是為胎兒的染色體異常所致:萎縮卵,葡萄胎=是萎縮性胚胎。至於萎縮卵發生的原因多為胚胎本身的問題
江西發現一例人類罕見異常染色體,此例染色體經實驗鑒定,屬世界首次發現。那麼,這異常染色體是怎麼形成呢?
江西省婦幼保健院近日從一名3歲男孩身上發現一例人類罕見異常染色體核型:46,XY,dup(4)(p12p16)。據醫院產前診斷中心主管技師李宇中介紹,這一異常染色體經中國醫學遺傳學國家重點實驗室鑒定,屬世界首次發現。
李宇中分析認為,這一染色體可能是在減數分裂時,同源染色體之間非對等交換或染色單體之間非對等交換形成重複片段而產生。根據基因的致病性程度,可能會導致患者先天性的非進行性智力低下,生長發育遲緩,並伴有五官、四肢、皮紋和內臟等方面的多發畸形癥狀。

基本信息
- 中文名
- 染色體異常
- 顯狀部位
- 全身